US20080139802A1 - Preparation of nucleosides ribofuranosyl pyrimidines - Google Patents
Preparation of nucleosides ribofuranosyl pyrimidines Download PDFInfo
- Publication number
- US20080139802A1 US20080139802A1 US11/973,748 US97374807A US2008139802A1 US 20080139802 A1 US20080139802 A1 US 20080139802A1 US 97374807 A US97374807 A US 97374807A US 2008139802 A1 US2008139802 A1 US 2008139802A1
- Authority
- US
- United States
- Prior art keywords
- aryl
- methyl
- afford
- contacting
- fluoro
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Abandoned
Links
- 0 *C1(C)C(=O)O[C@H](CC)[C@H]1C.*C1(C)C(C)O[C@H](CC)[C@H]1C.*C1(C)C(n2ccc(N)nc2=O)O[C@H](CC)[C@H]1C.C.C[Si](C)(C)Oc1ccnc(O[Si](C)(C)C)n1 Chemical compound *C1(C)C(=O)O[C@H](CC)[C@H]1C.*C1(C)C(C)O[C@H](CC)[C@H]1C.*C1(C)C(n2ccc(N)nc2=O)O[C@H](CC)[C@H]1C.C.C[Si](C)(C)Oc1ccnc(O[Si](C)(C)C)n1 0.000 description 3
- GSYVWKRRDIIBQG-RZXYLBDISA-N C=CC[C@H]1OC(=O)[C@](C)(F)[C@@H]1C=C.C=CC[C@H]1OC(Cl)[C@](C)(F)[C@@H]1C=C.C=CC[C@H]1O[C@@H](N2C=CC(NC(=O)C3=CC=CC=C3)=NC2=O)[C@](C)(F)[C@@H]1C=C.I.II.I[IH]I Chemical compound C=CC[C@H]1OC(=O)[C@](C)(F)[C@@H]1C=C.C=CC[C@H]1OC(Cl)[C@](C)(F)[C@@H]1C=C.C=CC[C@H]1O[C@@H](N2C=CC(NC(=O)C3=CC=CC=C3)=NC2=O)[C@](C)(F)[C@@H]1C=C.I.II.I[IH]I GSYVWKRRDIIBQG-RZXYLBDISA-N 0.000 description 2
- NYPIRLYMDJMKGW-VPCXQMTMSA-N C[C@@]1(F)[C@H](O)[C@@H](CO)O[C@H]1N1C=CC(N)=NC1=O Chemical compound C[C@@]1(F)[C@H](O)[C@@H](CO)O[C@H]1N1C=CC(N)=NC1=O NYPIRLYMDJMKGW-VPCXQMTMSA-N 0.000 description 2
- SSZGNAFRQIFYTO-NKKHWOTISA-N C.CC(=O)OC1O[C@H](COC(=O)C2=CC=CC=C2)[C@@H](OC(=O)C2=CC=CC=C2)[C@@]1(C)F.C[C@@]1(F)[C@H](O)[C@@H](CO)O[C@H]1N1C=CC(N)=NC1=O.C[C@@]1(F)[C@H](OC(=O)C2=CC=CC=C2)[C@@H](COC(=O)C2=CC=CC=C2)O[C@H]1N1C=CC(NC(=O)C2=CC=CC=C2)=NC1=O.C[C@]1(F)C(=O)O[C@H](COC(=O)C2=CC=CC=C2)[C@H]1OC(=O)C1=CC=CC=C1.C[C@]1(F)[C@@H](N2C=CC(NC(=O)C3=CC=CC=C3)=NC2=O)O[C@H](COC(=O)C2=CC=CC=C2)[C@H]1OC(=O)C1=CC=CC=C1.C[Si](C)(C)OC1=NC=CC(NC(=O)C2=CC=CC=C2)=N1 Chemical compound C.CC(=O)OC1O[C@H](COC(=O)C2=CC=CC=C2)[C@@H](OC(=O)C2=CC=CC=C2)[C@@]1(C)F.C[C@@]1(F)[C@H](O)[C@@H](CO)O[C@H]1N1C=CC(N)=NC1=O.C[C@@]1(F)[C@H](OC(=O)C2=CC=CC=C2)[C@@H](COC(=O)C2=CC=CC=C2)O[C@H]1N1C=CC(NC(=O)C2=CC=CC=C2)=NC1=O.C[C@]1(F)C(=O)O[C@H](COC(=O)C2=CC=CC=C2)[C@H]1OC(=O)C1=CC=CC=C1.C[C@]1(F)[C@@H](N2C=CC(NC(=O)C3=CC=CC=C3)=NC2=O)O[C@H](COC(=O)C2=CC=CC=C2)[C@H]1OC(=O)C1=CC=CC=C1.C[Si](C)(C)OC1=NC=CC(NC(=O)C2=CC=CC=C2)=N1 SSZGNAFRQIFYTO-NKKHWOTISA-N 0.000 description 1
- JTSMXYVKNZUCQW-FGHNCNNWSA-N C=CC[C@H]1OC(=O)[C@](C)(F)[C@@H]1C=C.C=CC[C@H]1OC(Cl)[C@](C)(F)[C@@H]1C=C.II.I[IH]I Chemical compound C=CC[C@H]1OC(=O)[C@](C)(F)[C@@H]1C=C.C=CC[C@H]1OC(Cl)[C@](C)(F)[C@@H]1C=C.II.I[IH]I JTSMXYVKNZUCQW-FGHNCNNWSA-N 0.000 description 1
- DORYKAJJQMJFDN-RPAKWOLASA-N CC1(C)OC[C@H]([C@@H](O)[C@H](O)[C@H]2COC(C)(C)O2)O1.CCOC(=O)/C(C)=C/[C@H]1COC(C)(C)O1.CCOC(=O)[C@@](C)(O)[C@H](O)[C@H]1COC(C)(C)O1.CCOC(=O)[C@@]1(C)OS(=O)(=O)O[C@@H]1[C@H]1COC(C)(C)O1.CC[C@H]1OC(=O)[C@](C)(F)[C@@H]1C.CC[C@H]1OC(Cl)[C@](C)(F)[C@@H]1OC(=O)C1=CC=CC=C1.CC[C@H]1O[C@@H](N2C=CC(C)=NC2=O)[C@](C)(F)[C@@H]1C.C[Si](C)(C)OC1=NC=CC(NC(=O)C2=CC=CC=C2)=N1 Chemical compound CC1(C)OC[C@H]([C@@H](O)[C@H](O)[C@H]2COC(C)(C)O2)O1.CCOC(=O)/C(C)=C/[C@H]1COC(C)(C)O1.CCOC(=O)[C@@](C)(O)[C@H](O)[C@H]1COC(C)(C)O1.CCOC(=O)[C@@]1(C)OS(=O)(=O)O[C@@H]1[C@H]1COC(C)(C)O1.CC[C@H]1OC(=O)[C@](C)(F)[C@@H]1C.CC[C@H]1OC(Cl)[C@](C)(F)[C@@H]1OC(=O)C1=CC=CC=C1.CC[C@H]1O[C@@H](N2C=CC(C)=NC2=O)[C@](C)(F)[C@@H]1C.C[Si](C)(C)OC1=NC=CC(NC(=O)C2=CC=CC=C2)=N1 DORYKAJJQMJFDN-RPAKWOLASA-N 0.000 description 1
- YXYHCOZXCNCWTC-MZKQUKJHSA-N CCOC(=O)/C(C)=C/C1COC(C)(C)O1.CCOC(=O)[C@@](C)(O)[C@H](O)C1COC(C)(C)O1 Chemical compound CCOC(=O)/C(C)=C/C1COC(C)(C)O1.CCOC(=O)[C@@](C)(O)[C@H](O)C1COC(C)(C)O1 YXYHCOZXCNCWTC-MZKQUKJHSA-N 0.000 description 1
- QRXSGYQUPUIBAC-VBMDWVFASA-M CCOC(=O)/C(C)=C/[C@H]1COC(C)(C)O1.CCOC(=O)[C@@](C)(O)[C@H](O)[C@H]1COC(C)(C)O1.[V].[V]I Chemical compound CCOC(=O)/C(C)=C/[C@H]1COC(C)(C)O1.CCOC(=O)[C@@](C)(O)[C@H](O)[C@H]1COC(C)(C)O1.[V].[V]I QRXSGYQUPUIBAC-VBMDWVFASA-M 0.000 description 1
- FKEGWAWRZKIAMV-VNTKNQDVSA-I CCOC(=O)[C@@]1(C)OS(=O)(=O)O[C@@H]1[C@H]1COC(C)(C)O1.I[V](I)I.I[V]I Chemical compound CCOC(=O)[C@@]1(C)OS(=O)(=O)O[C@@H]1[C@H]1COC(C)(C)O1.I[V](I)I.I[V]I FKEGWAWRZKIAMV-VNTKNQDVSA-I 0.000 description 1
- PQQNMQIWFDLFFF-CZJHLHDVSA-K CCOC(=O)[C@@]1(C)OS(=O)O[C@@H]1[C@H]1COC(C)(C)O1.I[V]I.[V]I Chemical compound CCOC(=O)[C@@]1(C)OS(=O)O[C@@H]1[C@H]1COC(C)(C)O1.I[V]I.[V]I PQQNMQIWFDLFFF-CZJHLHDVSA-K 0.000 description 1
- SMUVCPHULLHHQL-VOSJYQQWSA-K CCOC(=O)[C@](C)(F)[C@H](OS(=O)(=O)[O-])[C@H]1COC(C)(C)O1.CC[NH+](CC)CC.C[C@]1(F)C(=O)O[C@H](CO)[C@H]1O.I[V](I)I Chemical compound CCOC(=O)[C@](C)(F)[C@H](OS(=O)(=O)[O-])[C@H]1COC(C)(C)O1.CC[NH+](CC)CC.C[C@]1(F)C(=O)O[C@H](CO)[C@H]1O.I[V](I)I SMUVCPHULLHHQL-VOSJYQQWSA-K 0.000 description 1
- KPZDSZTWCLVXRS-ATRFCDNQSA-N CC[C@H]1OC(=O)[C@](C)(F)[C@@H]1C Chemical compound CC[C@H]1OC(=O)[C@](C)(F)[C@@H]1C KPZDSZTWCLVXRS-ATRFCDNQSA-N 0.000 description 1
- NOZMNZFISHLGQT-ZDNAGBJOSA-N COCCOOCCOC.C[C@@]1(F)[C@H](OC(=O)C2=CC=CC=C2)[C@@H](COC(=O)C2=CC=CC=C2)O[C@H]1N1C=CC(NC(=O)C2=CC=CC=C2)=NC1=O.C[C@]1(F)C(Cl)O[C@H](COC(=O)C2=CC=CC=C2)[C@H]1OC(=O)C1=CC=CC=C1.[Na][AlH2] Chemical compound COCCOOCCOC.C[C@@]1(F)[C@H](OC(=O)C2=CC=CC=C2)[C@@H](COC(=O)C2=CC=CC=C2)O[C@H]1N1C=CC(NC(=O)C2=CC=CC=C2)=NC1=O.C[C@]1(F)C(Cl)O[C@H](COC(=O)C2=CC=CC=C2)[C@H]1OC(=O)C1=CC=CC=C1.[Na][AlH2] NOZMNZFISHLGQT-ZDNAGBJOSA-N 0.000 description 1
- MFXWNWXGASXTHC-VFZAHCDKSA-M C[C@@]1(F)[C@H](O)[C@@H](CO)O[C@H]1N1C=CC(N)=NC1=O.I.[V]I Chemical compound C[C@@]1(F)[C@H](O)[C@@H](CO)O[C@H]1N1C=CC(N)=NC1=O.I.[V]I MFXWNWXGASXTHC-VFZAHCDKSA-M 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H5/00—Compounds containing saccharide radicals in which the hetero bonds to oxygen have been replaced by the same number of hetero bonds to halogen, nitrogen, sulfur, selenium, or tellurium
- C07H5/02—Compounds containing saccharide radicals in which the hetero bonds to oxygen have been replaced by the same number of hetero bonds to halogen, nitrogen, sulfur, selenium, or tellurium to halogen
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H1/00—Processes for the preparation of sugar derivatives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H19/00—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof
- C07H19/02—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof sharing nitrogen
- C07H19/04—Heterocyclic radicals containing only nitrogen atoms as ring hetero atom
- C07H19/06—Pyrimidine radicals
Abstract
The present process provides an improved method for converting 2′-deoxy-2′-fluoro-2′-methyl-D-ribonolactones derivatives to 3-fluoro-3-methyl-2-chlorofuran compounds which are useful for the synthesis of nucleosides and improved processes for the synthesis of the D-ribonolactone compounds.
Description
- This application is claims benefit of U.S. Provisional Application No. 60/850,962, filed Oct. 10, 2006, which is hereby incorporated by reference in its entirety.
- The present invention relates to an improved process for the transformation of an O-acyloxy-substituted (R)-5-methyl-dihydro-furan-2-one derivative into an O-acyloxy-substituted (R)-2-chloro-5-methyl-tetrahydro-furan. The present invention further provides a method of synthesis of cytosine and uridine derivatives utilizing the O-acyloxy-substituted (R)-2-chloro-5-methyl-tetrahydro-furan. The reaction sequence provides an improved process for the preparation of 4-amino-1-((2R,3R,4R,5R)-3-fluoro-4-hydroxy-5-hydroxymethyl-3-methyl-tetrahydro-furan-2-yl)-1H-pyrimidin-2-one (18) which is a potent inhibitor of Hepatitis C Virus (HCV) NS5B polymerase. Another feature of the present invention is improved procedures for the synthesis of key lactone intermediate 10 utilized in the synthesis of 18.
- The successful use of modified nucleosides to treat viral and neoplastic diseases has prompted continuing interest in identifying new therapeutically useful nucleosides. The synthesis of pyrimidine nucleosides has been reviewed (T. Ueda, “Synthesis and Reactions of Pyrimidine Nucleosides”, in Chemistry of Nucleosides and Nucleotides, L. B. Townsend Ed.; Plenum Press: New York, Year, vol. 1, pp 1-95; H. Vorbrüggen, “Methods of Nucleoside Synthesis”. In Nucleoside Analogues; R. T. Walker, E. De Clercq and F. Eckstein, Eds.; NATO Advanced Study Institutes Series, Series A, (Life Sciences); Plenum Press: New York, 1979, A26, pp. 35-69). A critical step in the synthesis of nucleosides is often the condensation of the nucleobase and carbohydrate to form the N-glycosidic bond. 2-Deoxy-nucleosides afford unique problems because they lack a 2-hydroxy substituent that provides 1,2-anchiomeric assistance facilitating stereoselective β-glycosylation at the adjacent anomeric carbon.
-

- Traditional carbohydrate leaving groups on the carbohydrate moiety (X in formula 4) include halides, acetates and methanesulfonyloxy groups. Vorbrüggen, et al. (J. Org. Chem. 1976 41:2084) disclosed improvements in the glycosylation reaction of heterocyclic bases and demonstrated that nucleosides may be obtained from the Lewis-acid catalyzed reaction of a peracylated carbohydrate and silylated heterocycles in a solvent such as DCE or MeCN.
- After protection of the hydroxyl groups, the lactone is reduced to the corresponding lactol. A preferred reducing agent is DIBAL-H wherein the reaction temperature range between about −100° to −20° C. The reaction conditions must be carefully controlled to avoid lactol ring opening and further reduction. Other metal hydrides, such as the widely used lithium aluminum hydride, can also be used for the reduction, but it is necessary to keep the temperature low and to ensure that all the hydride is destroyed before an elevation of the temperature occurs which is difficult in a large scale process. Accordingly, a solvent with a very low freezing point must be used in the reduction step. Toluene is convenient, but other solvents can be used, including ethers such as diethyl ether, THF and DME and the like.
- After reduction to the lactol, an appropriate leaving group must be introduced into the 1-position of the carbohydrate to allow for the efficient incorporation of the heterocyclic base. Useful activated sugars include alkylsulfonyloxy (4, X═OSO2Me), alkanoyloxy (4, X═OCOMe) and halo (4, X=Cl or Br) substituted sugars. Alkanoyloxy substituted sugars can be prepared by acylation of the lactol with an acylating agent such as acetic anhydride or acetyl chloride and the like in the presence of an equivalent amount of a suitable acid scavenger such as a trialkylamine or pyridine, e.g., TEA or DIPEA and the like. In U.S. Pat. No. 4,526,988 published Jul. 2, 1985, L. W. Hertel teaches methanesulfonyloxy substituted sugars are very effective glycosylating agents which are prepared analogously using methanesulfonyl chloride in place of the acylating agent. Other sulfonyl leaving groups are provided in the same way by reaction with the appropriate sulfonyl halide.
- When a chloro or bromo leaving group is to be used, the halo derivative is commonly prepared from the lactol in a two-step sequence. The 1-acetoxy derivative is prepared by reacting the lactol with acetic anhydride, or another source of acetyl groups, in the presence of one or more equivalents an acid scavenger. The acetate group is then displaced with a halide by treatment with gaseous hydrogen bromide or hydrogen chloride, at a low temperature such as about −50° C. to about 0° C. Since the gaseous hydrogen halide tends to remove the protecting groups, especially silyl protecting groups, it is necessary to operate this step at quite a low temperature and to add the hydrogen halide slowly in small increments.
- Vorbrüggen has shown that the coupling of the nucleobase and the sugar moiety can be efficiently catalyzed with Lewis acids such as stannic chloride, trimethylsilyl trifluoromethanesulfonate and trimethylsilyl perchlorate. (H. Vorbrüggen and K. Krolikiewicz, Angew Chem. Int. Ed. Eng. 14, 421:1975; H. Vorbrüggen et al., Chem. Ber. 1981, 114:1234)
- In US 20050009737 published Jan. 13, 2005, J. Clark discloses fluoro-nucleoside derivatives that inhibit Hepatitis C Virus (HCV) NS5B polymerase. In particular, 4-amino-1-((2R,3R,4R,5R)-3-fluoro-4-hydroxy-5-hydroxymethyl-3-methyl-tetrahydro-faran-2-yl)-1H-pyrimidin-2-one (18) was a particularly potent inhibitor of HCV polymerase as well as the polymerase of other Flaviviridae.
-

- In WO2006/012440 published Feb. 2, 2006, P. Wang et al disclose processes for the preparation of 18. Introduction of the cytosine is carried out utilizing the Vorbruggen protocol. In US 20060122146 published Jun. 8, 2006, B.-K. Chun et al. disclose and improved procedures for the preparation of the 2-methyl-2-fluoro-lactone 10. In the latter disclosure the nucleobase is glycosylated by reacting with ribofuranosyl acetate which is prepared by reduction of 10 with LiAlH(O-tert-Bu)3 followed by acetylaton of the intermediate lactol which was treated with an O-trimethylsilyl N4-benzoylcytosine in the presence of SnCl4 to afford the O,O,N-tribenzoylated nucleoside.
- The current invention affords an improved process for the synthesis of nucleosides which comprises the steps of:
-

-
- (i) contacting an (aryl)alkanoic acid (2R,3R,4R)-2-(aryl)alkanoyloxymethyl-4-fluoro-4-methyl-5-oxo-tetrahydro-furan-3-yl ester (II; R=aryl or alkyl) with RED-AL/2,2,2-trifluoroethanol (TFE) (1:1) in DCM/PhMe (6:1) at a temperature of −10 to −5° C.;
- (ii) contacting the mixture with a chlorinating agent selected from sulfuryl chloride, thionyl chloride or phosphorus oxychloride and tetrabutylammonium bromide to afford an (aryl)alkanoic acid (2R,3R,4R)-2-(aryl)alkanoyloxy methyl-5-chloro-4-fluoro-4-methyl-tetrahydro-furan-3-yl ester (III; R=aryl or alkyl); and,
- (iii) contacting III with N-(2-trimethylsilanyloxy-pyrimidin-4-yl)-benzamide, SnCl4 and PhCl to afford an (aryl)alkanoic acid (2R,3R,4R,5R)-3-(aryl)alkanoyloxy-5-(4-benzoylamino-2-oxo-2H-pyrimidin-1-yl)-4-fluoro-4-methyl-tetrahydro-furan-2-ylmethyl ester (I; R=aryl or alkyl).
- The current invention further provides for improved procedures for the stereopecific hydroxylation of (E)-3-((S)-2,2-dimethyl-[1,3]dioxolan-4-yl)-2-methyl-acrylic acid ethyl ester (22), the formation of the cyclic sulfate 26 and the ring-opening of the cyclic sulfate with a fluoride nucleophile and hydrolysis, lactonization and derivatization to afford the required lactone II (R=Ph). The latter three steps afford an efficient scalable process to prepare the requisite lactone (II).
- While nucleosides have proven valuable chemotherapeutic agents, the large-scale synthesis of nucleosides remains a challenging task. New efficient and cost effective processes for the synthesis of nucleosides and key precursors are essential to provide commercially viable new drug entities. In one embodiment of the present invention there is provided a process for conversion of a substituted γ-lactone II (wherein R is an aryl or alkyl group) to the corresponding 2-chlorofuran by treating II with sodium bis-(2-methoxyethoxy)(2,2,2-trifluoro-ethoxy)aluminum hydride and contacting the resulting alcoholate with a chlorinating agent selected from sulfuryl chloride, thionyl chloride or phosphorus oxychloride in the presence of catalytic quantities of tetrabutylammonium bromide. The resulting substituted 2-chlorofuran derivative is condensed with O-trimethylsilyl-N4-benzoylcytosine in the presence of a Lewis acid to afford the perbenzoylated nucleoside. One skilled in the art would appreciate that the process is sufficiently general that the other ester and amides can be used as protecting groups in place of the benzoic acid derivatives without departing from the spirit of the invention.
- In another embodiment of the present invention there is provided a process for conversion of a substituted γ-lactone II (wherein R is an aryl or alkyl group) to the corresponding 2-chlorofuran by treating II with sodium bis-(2-methoxyethoxy)(2,2,2-trifluoro-ethoxy)aluminum hydride and contacting the resulting alcoholate with a chlorinating agent selected from sulfuryl chloride, thionyl chloride or phosphorus oxychloride in the presence of catalytic quantities of tetraalkylammonium salt or phosphonium salt. The resulting substituted 2-chlorofuran derivative is condensed with O-trimethylsilyl-N4-benzoylcytosine in the presence of a Lewis acid to afford the perbenzoylated nucleoside. One skilled in the art would appreciate that the process is sufficiently general that the other ester and amides can be used as protecting groups in place of the benzoic acid derivatives without departing from the spirit of the invention.
- In another embodiment of the present invention there is provided a process for conversion of a substituted γ-lactone II (wherein R is an aryl or alkyl group) to the corresponding 2-chlorofuran by treating II with sodium bis-(2-methoxyethoxy)(2,2,2-trifluoro-ethoxy)aluminum hydride and contacting the resulting alcoholate with sulfuryl chloride in the presence of catalytic quantities of tetrabutylammonium bromide. The resulting substituted 2-chlorofuran derivative is condensed with O-trimethylsilyl-N4-benzoylcytosine in the presence of a Lewis acid to afford the perbenzoylated nucleoside. One skilled in the art would appreciate that the process is sufficiently general that the other ester and amides can be used as protecting groups in place of the benzoic acid derivatives without departing from the spirit of the invention.
- In another embodiment of the present invention the nucleoside is prepared from the lactone II wherein R is an aryl or alkyl group as described in the summary of the invention and the resulting acylated nucleoside is hydrolyzed under basic conditions to afford the unprotected cytidine nucleoside 18.
- In yet another embodiment of the present invention the lactone 10 is converted to the acylated nucleoside 14 utilizing O-trimethylsilyl-N4-benzoylcytosine and the benzoyl groups are hydrolyzed to afford 18.
- In still another embodiment of the present invention the olefin 22 is stereospecifically dihydroxylated with aqueous sodium permanganate, ethylene glycol, sodium bicarbonate and acetone at −20 to 0° C., then quenched with aqueous sodium bisulfite. In this embodiment the resulting diol is cyclized with thionyl chloride to afford the cyclic sulfite 36 which is oxidized to the cyclic sulfate with NaOCl in the presence of MeCN to afford the cyclic sulfate 26. The cyclic sulfate is exposed to triethylamine-trihydrofluoride/TEA and the intermediate fluorohydrin sulfate 38 is decomposed in the presence of BaCl2 under aqueous acidic conditions to afford the lactone 28.
- In another embodiment of the present invention there is provided a process for conversion of a substituted peracylated (4S,5R)-4-hydroxy-5-hydroxymethyl-dihydro-furan-2-one II (wherein R is an aryl or alkyl group) to the corresponding 2-chlorofuran III by treating II with sodium bis-(2-methoxyethoxy)(2,2,2-trifluoro-ethoxy)aluminum hydride then treating the reaction mixture with sulfuryl chloride, phosphorus oxychloride or thionyl chloride.
- In another embodiment of the present invention there is provided a process for conversion of a substituted peracylated (4S,5R)-4-hydroxy-5-hydroxymethyl-dihydro-furan-2-one II (wherein R is an aryl or alkyl group) to the corresponding 2-chlorofuran III by treating II with sodium bis-(2-methoxyethoxy)(2,2,2-trifluoro-ethoxy)aluminum hydride then treating the reaction mixture with sulfuryl chloride.
- The phrase “a” or “an” entity as used herein refers to one or more of that entity; for example, a compound refers to one or more compounds or at least one compound. As such, the terms “a” (or “an”), “one or more”, and “at least one” can be used interchangeably herein.
- The term (aryl)alkanoic acid as used herein refers to a group RCO2H wherein R is either alkyl or aryl as those terms are defined herein. Correspondingly the term (aryl)alkanoic ester refers to a group RCO2R′ where R is either alkyl or aryl. Most typically the R′ represents the 3′ and/or 5′ position(s) of a ribose ring. The terms “(aryl)alkanoyl” and “(aryl)alkanoyloxy” refer to the groups RCO— and RCOO— respectively, where R is as described previously. The term “(aryl)alkanoyloxymethyl” group refers a group RCOOCH2— where R is as described previously.
- The term “alkyl” as used herein denotes an unbranched or branched chain, saturated, monovalent hydrocarbon residue containing 1 to 10 carbon atoms.
- The term “aryl” as used herein refers to phenyl group.
- An O-acyloxy-(R)-5-methyl-dihydro-furan-2-one derivative refers to a lactone of formula (i). An O-acylated-(R)-2-chloro-5-methyl-tetrahydro-furan derivative refers to a compound of
-

- formula (ii) wherein R is aryl or alkyl and R′ and R″ are independently hydrogen, halogen or C1-3 alkyl.
- As used herein, the term “treating”, “contacting” or “reacting” when referring to a chemical reaction means to add or mix two or more reagents under appropriate conditions to produce the indicated and/or the desired product. It should be appreciated that the reaction which produces the indicated and/or the desired product may not necessarily result directly from the combination of two reagents which were initially added, i.e., there may be one or more intermediates which are produced in the mixture which ultimately leads to the formation of the indicated and/or the desired product.
- The term “acylating agent” as used herein refers to either an anhydride, acid halide or an activated derivative of an N-protected alpha amino acid. The term “anhydride” as used herein refers to compounds of the general structure RC(O)—O—C(O)R wherein R is aryl or alkyl as defined herein. The term “acid halide” as used herein refers to compounds of the general structure RC(O)X wherein X is a halogen and wherein R is aryl or alkyl as defined herein. The term “activated derivative” is as defined below.
- The term “activated derivative” of a compound as used herein refers to a transient reactive form of the original compound which renders the compound active in a desired chemical reaction, in which the original compound is only moderately reactive or non-reactive. Activation is achieved by formation of a derivative or a chemical grouping within the molecule with a higher free energy content than that of the original compound, which renders the activated form more susceptible to react with another reagent. In the context of the present invention activation of the carboxy group is of particular importance and corresponding activating agents or groupings which activate the carboxy group are described in more detail below.
- The term “amino-protecting group” as used herein refers to a protecting group that preserves a reactive amino group that otherwise would be modified by certain chemical reactions. The definition includes the formyl group or lower alkanoyl groups with 2 to 4 carbon atoms, in particular the acetyl or propionyl group, the trityl or substituted trityl groups such as the monomethoxy-trityl group, dimethoxytrityl groups such as the 4,4′-dimethoxytrityl, the trichloroacetyl group, the trifluoroacetyl group, the silyl group, the phthalyl group, and N-urethanes. Preferred amino-protecting groups are N-urethanes such as the N-benzyloxycarbonyl group (cbz) derived from benzylchlorocarbonate or N-alkoxycarbonyl group, e.g. tert-butoxycarbonyl which is prepared by reaction with di(t-butyl)dicarbonate.
- The term “hydroxyl protecting group” or “alcohol protecting group” means a protecting group that preserves a hydroxy group that otherwise would be modified by certain chemical reactions. Reagents and protocols for to introduce and remove amino- and hydroxy-protecting groups are well known and have been reviewed in numerous texts (e.g., T. W. Greene and P. G. M. Wuts, Protective Groups in Organic Synthesis, 3rd edition, John Wiley & Sons, New York, 1999, and Harrison and Harrison et al., Compendium of Synthetic Organic Methods, Vols. 1-8 John Wiley and Sons, 1971-1996).
- The term “nucleobase” as used here refers to the nitrogen contain heterocyclic moiety linked to C1 of the carbohydrate.
- Commonly used abbreviations include: acetyl (Ac), azo-bis-isobutyrylnitrile (AIBN), atmospheres (Atm), tert-butoxycarbonyl (Boc), di-tert-butyl pyrocarbonate or boc anhydride (BOC2O), benzyl (Bn), butyl (Bu), benzyloxycarbonyl (CBZ or Z), carbonyl diimidazole (CDI), diethylaminosulfur trifluoride (DAST), N,N′-dicyclohexylcarbodiimide (DCC), 1,2-dichloroethane (DCE), dichloromethane (DCM), di-iso-butylaluminumhydride (DIBAL or DIBAL-H), di-iso-propylethylamine (DIPEA), N,N-dimethyl acetamide (DMA), 4-N,N-dimethylaminopyridine (DMAP), N,N-dimethylformamide (DMF), dimethyl sulfoxide (DMSO), 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDCI), ethyl (Et), ethyl acetate (EtOAc), ethanol (EtOH), 2-ethoxy-2H-quinoline-1-carboxylic acid ethyl ester (EEDQ), diethyl ether (Et2O), O-(7-Azabenzotriazole-1-yl)-N,N,N′N′-tetramethyluronium hexafluorophosphate acetic acid (HATU), acetic acid (HOAc), 1-N-hydroxybenzotriazole (HOBt), high pressure liquid chromatography (HPLC), lithium hexamethyl disilazane (LiHMDS), methanol (MeOH), melting point (mp), MeSO2— (mesyl or Ms), methyl (Me), acetonitrile (MeCN), m-chloroperbenzoic acid (MCPBA), mass spectrum (ms), methyl t-butyl ether (MTBE), N-bromosuccinimide (NBS), N-chlorosuccinimide (NCS), N-methylmorpholine (NMM), N-methylpyrrolidone (NMP), phenyl (Ph), propyl (Pr), iso-propyl (i-Pr), pounds per square inch (psi), pyridine (pyr), room temperature (rt or RT), tert-butyldimethylsilyl or t-BuMe2Si (TBDMS), triethylamine (TEA or Et3N), 2,2,6,6-tetramethylpiperidine 1-oxyl (TEMPO), triflate or CF3SO2— (Tf), trifluoroacetic acid (TFA), thin layer chromatography (TLC), tetrahydrofuran (THF), trimethylsilyl or Me3Si (TMS), p-toluenesulfonic acid monohydrate (TsOH or pTsOH), 4-Me-C6H4SO2— or tosyl (Ts). Conventional nomenclature including the prefixes normal (n), iso (i-), secondary (sec-), tertiary (tert-) and neo have their customary meaning when used with an alkyl moiety. (J. Rigaudy and D. P. Klesney, Nomenclature in Organic Chemistry, IUPAC 1979 Pergamon Press, Oxford.).
- In general, the nomenclature used in this Application is based on AUTONOM™ v.4.0, a Beilstein Institute computerized system for the generation of IUPAC systematic nomenclature. If there is a discrepancy between a depicted structure and a name given that structure, the depicted structure is to be accorded more weight. In addition, if the stereochemistry of a structure or a portion of a structure is not indicated with, for example, bold or dashed lines, the structure or portion of the structure is to be interpreted as encompassing all stereoisomers of it.
- Some compounds in following schemes are depicted with generalized substituents; however, one skilled in the art will immediately appreciate that the nature of the R groups can varied to afford the various compounds contemplated in this invention. Moreover, the reaction conditions are exemplary and alternative conditions are well known. The reaction sequences in the following examples are not meant to limit the scope of the invention as set forth in the claims.
- The present invention provides an improved process for the preparation of 18. Lithium tris-(tert-butoxy)-aluminum hydride and sodium bis-2-methoxyethoxy)aluminum hydride are illustrative of hydride reagents which have been found to modulate the reactivity of LiAlH4 and to improve solubility in organic solvents and increase stability. Sodium bis-(2-methoxyethoxy)(1,1,1,3,3,3-hexafluoro-2-propoxy)aluminum hydride has also been reported to be a useful reducing agent (S. Harashima et al., Tetrahedron 1991 47(16/17):2773-2784). The reduction of 10 with sodium bis-(2-methoxyethoxy)(2,2,2-trifluoro-ethoxy)aluminum hydride and contacting the resulting aluminate salt with sulfuryl chloride and tetrabutylammonium bromide afforded the ribosyl chloride 30 directly. Contacting 30 with silylated N4-benzoylcytosine (32) in the presence of SnCl4 or a Lewis acid affords the tribenzoylated nucleoside 14 which is readily hydrolyzed to the nucleoside with catalytic quantities of sodium methoxide in methanol. It would be readily apparent to one skilled in the art that although the process is exemplified used the N4-benzoyl moiety as an N protecting group, many N-protecting groups are know and the choice of a specific group is merely a matter of convenience.
- The present process as described in SCHEME A and the following examples contain numerous improvements which have resulted in higher yields of the desired nucleoside. The asymmetric hydroxylation of 22 was discovered to be best carried out with sodium permanganate in the presence of ethylene glycol, sodium bicarbonate in acetone which afforded the diol in 60-64% on pilot plant scale. The sodium permanganate procedure avoids introduction of osmium into the process stream. Further more the stereospecific hydroxylation can be accomplished without using an expensive chiral ligand. The requisite olefin is prepared from (1S,2S)-1,2-bis-((R)-2,2-dimethyl-[1,3]dioxolan-4-yl)-ethane-1,2-diol (20) (C. R. Schmid and J. D. Bryant, Org. Syn. 1995 72:6-13) by oxidative cleavage of the diol and treating the resulting aldehyde with 2-(triphenyl-λ5-phosphanylidene)-propionic acid ethyl ester to afford 22.
-

- (i) NaIO4, NaHCO3, DCM; (ii) MeC(═PPh3)CO2Et; (iii) acetone-NaMnO4 (aq), ethylene glycol, NaHCO3, −10 to 0° C.; aq. NaHSO3 (quench); (iv) i-PrOAc, MeCN, TEA, SOCl2; (v) i-PrOAc, MeCN, NaOCl; (vi) TEA-3HF, TEA; (vii) HCl (aq)-BaCl2-aq; (viii) (PhCO)2O, DMAP, MeCN, (ix) RED-AL/TFE (1:1), DCM; (x) SO2Cl2-TBAB, DCM; (xi) 32, SnCl4-PhCl; (xii) MeOH-MeONa
- Cyclic sulfates can be prepared by cyclization of a vicinal diol with thionyl chloride and oxidation of the resulting cyclic sulfite to the corresponding sulfate. Cyclic sulfates have been previously found to be useful synthetic intermediates. (Y. Gao and K. B. Sharpless, J. Am. Chem. Soc. 1988 110(22):7538-7539) It has now been found that the cyclic sulfates are stabilized during processing by the addition of a trialkylamine such as TEA or DIPEA at 2-10 mole % relative to the cyclic sulfate. Differential scanning calorimetry (DSC) showed a shift in the onset of decomposition from approximately 110 to 180° C. when 3.5 mole % of DIPEA was added to 26. Contacting 26 with triethylamine-trihydrofluoride/TEA affords the sulfated fluorohydrin which in the presence of water affords the fluorohyrin. It has now been found that an improved yield of 28 is obtained when BaCl2 is incorporated in the reaction mixture to scavenge the liberated sulfate. Under the acidic conditions concomitant hydrolysis of the acetonide liberates a triol which spontaneously cyclizes to the γ-lactone 28. Contacting 28 with benzoic anhydride and DMAP affords the dibenzoate of the lactone 10 which is used in the glycosylation step.
- The esters are alcohol protecting groups. Other protecting groups can also be employed successfully without departing from the invention. The lactone 28 can be fully protected with appropriate protection agents with an appropriate base in an appropriate solvent. Reaction conditions for the formation of esters are well known in the art. The potential protecting groups include, but are not limited to the following: methoxymethyl, methoxyethyl, benzyloxymethyl, ethoxymethyl, trityl, triethylsilyl, t-butyldimethylsilyl, t-butyldiphenylsilyl, acyl including acetyl, pivaloyl, benzoyl, toluoyl, 4-phenyl benzoyl, 2-, 3-, or 4-nitrobenzoyl, 2-, 3-, or 4-chlorobenzoyl, other substituted benzoyl. Useful bases include, but are not limited to the following list: imidazole, pyridine, DMAP, TEA, DIPEA, 1,4-diazabicyclo[2,2,2]octane. Useful solvents include, but are not limited to pyridine, DCM, chloroform, DCE, THF.
-
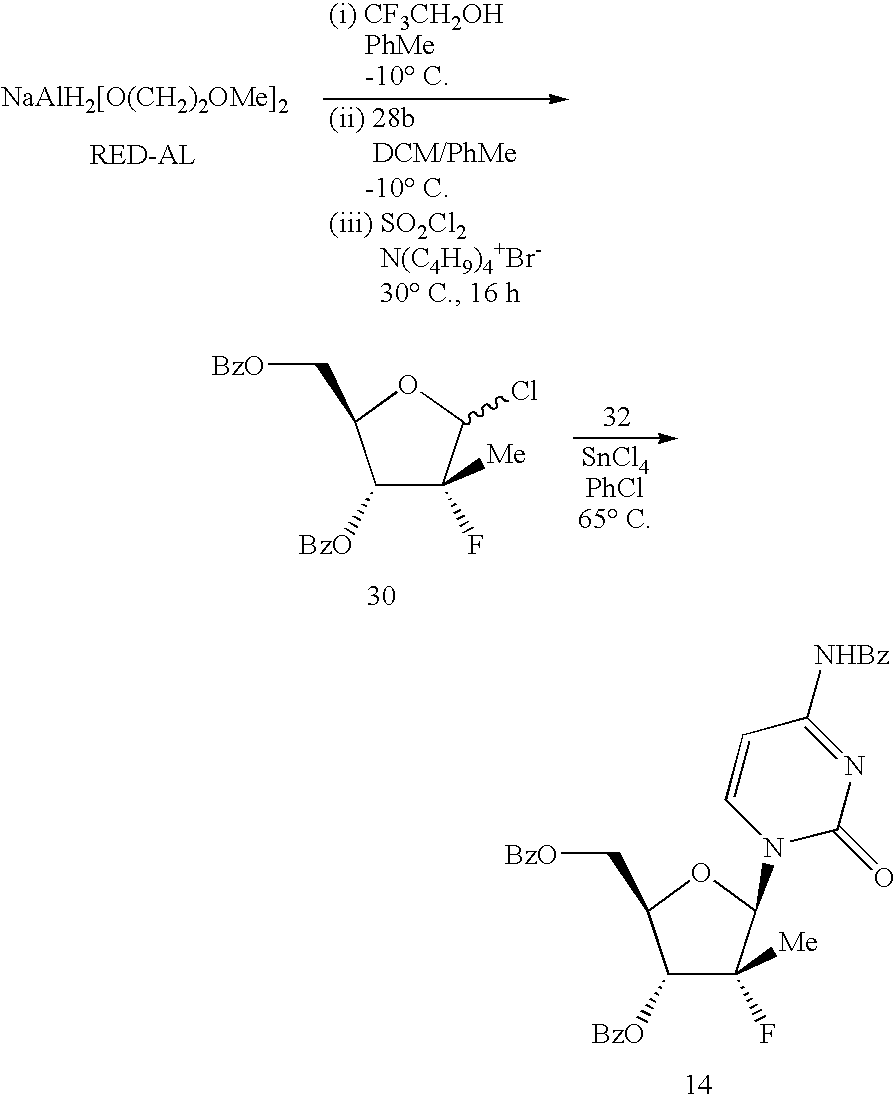
- Trifluoroethanol (4.08 kg) is added slowly to a cold solution (−15° C.) of RED-AL® solution (12.53 kg) and toluene (21.3 kg) while maintaining the reaction temperature at or below −10° C. After warming up to RT (ca. 20° C.), the modified RED-AL reagent mixture (30.1 kg out of the 37.6 kg prepared) is added slowly to a pre-cooled solution (−15° C.) of fluorolactone dibenzoate 10 (10 kg) in DCM (94.7 kg) while maintaining reaction temperature at or below −10° C. After reduction of the lactone (monitored by in-process HPLC), a catalytic amount of tetrabutylammonium bromide (90 g) is added to the reaction mixture. Sulfiiryl chloride (11.86 kg) is then added while maintaining reaction temperature at or below 0° C. The reaction mixture is then heated to 40° C. until formation of the chloride is complete (ca. 4 h) or warmed to RT (20-25° C.) and stirred over night (ca. 16 h). The reaction mixture is cooled to about 0° C., and water (100 L) is added cautiously while maintaining reaction temperature at or below 15° C. The reaction mixture is then stirred at RT for ca. 1 h to ensure hydrolytic decomposition of excess sulfuryl chloride and the phases are separated. The organic layer is washed with a dilute solution of citric acid (prepared by dissolving 15.5 kg of citric acid in 85 L of water) and then with dilute KOH solution (prepared by dissolving 15 kg of 50% KOH in 100 L of water). The organic phase is then concentrated and solvents are replaced with chlorobenzene (2×150 kg) via atmospheric replacement distillation. The resulting solution containing 30 is dried azeotropically.
- A suspension of N-benzoyl cytosine (8.85 kg), ammonium sulfate (0.07 kg) and hexamethyldisilazane (6.6 kg) in chlorobenzene (52.4 kg) is heated to reflux (ca. 135° C.) and stirred (ca. 1 h) until the mixture becomes a clear solution. The reaction mixture is then concentrated in vacuo to obtain 32 as a syrupy liquid. The anhydrous solution of 30 in chlorobenzene (as prepared) and stannic chloride (28.2 kg) is added to this concentrate. The reaction mixture is maintained at about 70° C. until the desired coupling reaction is complete (ca. 10 h) as determined by in-process HPLC. Upon completion, the reaction mixture is cooled to RT and diluted with DCM (121 kg). This solution is added to a suspension of solid NaHCO3 (47 kg) and CELITE® (9.4 kg) in DCM (100.6 kg). The resulting slurry is cooled to 10-15° C., and water (8.4 kg) is added slowly to quench the reaction mixture. The resulting suspension is very slowly (caution: gas evolution) heated to reflux (ca. 45° C.) and maintained for about 30 min. The slurry is then cooled to ca. 15° C. and filtered. The filter cake is repeatedly reslurried in DCM (4×100 L) and filtered. The combined filtrate is concentrated under atmospheric pressure (the distillate collected in the process is used for reslurrying the filter cake) until the batch temperature rises to about 90° C. and then allowed to cool slowly to about −5° C. The resulting slurry is aged for at least 2 h at −5° C. The precipitated product is filtered and washed with IPA (30 kg+20 kg), and oven-dried in vacuo at about 70° C. to afford 8.8 kg (57.3%) of 1-(2-deoxy-2-fluoro-2-methyl-3-5-O-dibenzoyl-β-D-ribofuranosyl)-N-4-benzoylcytosine (14, CAS Reg No. 817204-32-3) which was 99.3% pure.
-

- A slurry of 14 (14.7 kg) in MeOH (92.6 kg) is treated with catalytic amounts of methanolic sodium methoxide (0.275 kg). The reaction mixture is heated to ca. 50° C. and aged (ca. 1 h) until the hydrolysis is complete. The reaction mixture is quenched by addition of isobutyric acid (0.115 kg). The resulting solution is concentrated under moderate vacuum and then residual solvents are replaced with IPA (80 kg). The batch is distilled to a volume of ca. 50 L. The resulting slurry is heated to ca. 80° C. and then cooled slowly to ca. 5° C. and aged (ca. 2 h). The precipitated product is isolated by filtration, washed with IPA (16.8 kg) and dried in an oven at 70° C. in vacuo to afford 6.26 kg (88.9%) of 18 which assayed at 99.43% pure.
-

- A suspension of 22 (10 kg, CAS Reg. No. 81997-76-4), ethylene glycol (11.6 kg), solid NaHCO3 (11.8 kg) and acetone (150 L) is cooled to ca.-15° C. A solution of 36% aqueous NaMnO4 (19.5 kg) is charged slowly (over 4 h) to the suspension maintaining reaction temperature at or below −10° C. After stirring for 0.5 h at −10° C., an aliquot of the reaction mixture (ca. 5 mL) is quenched with 25% aqueous sodium bisulfite (ca. 15 mL). A portion of resulting slurry is filtered and submitted for GC analysis to check the progress of the reaction. When the reaction is complete, the reaction mixture is quenched by slow addition (over 40 min) of cooled (ca. 0° C.) 25% aqueous NaHSO3 (60 L). The temperature of the reaction mixture is allowed to reach 4° C. during the quench. CELITE® (ca. 2.5 kg) is then slurried in acetone (8 kg) and added to the dark brown reaction mixture. The resulting slurry is aged at RT to obtain light tan slurry. The slurry is filtered, and the filter cake is washed with acetone (3×39 kg). The combined filtrate is concentrated by vacuum distillation (vacuum approximately 24 inches of Hg; max pot temperature is 32° C.) to remove the acetone. The aqueous concentrate is extracted with EtOAc (3×27 kg), and the combined organic extracts were washed with water (25 L). The organic phase is then concentrated by atmospheric distillation and EtOAc is replaced with toluene. The volume of the batch is adjusted to ca. 20 L. Heptane (62 kg) is added and the batch cooled to ca. 27° C. to initiate crystallization. The batch is then cooled to −10° C. After aging overnight at −10° C., the product is filtered, washed with 10% toluene in heptane and dried at 50° C. under vacuum to afford 6.91 kg (59.5%) of 24 (CARN 81997-76-4) as a white crystalline solid.
-

- steps 1 & 2—A dry, clean vessel was charged with 24 (6.0 kg), isopropyl acetate (28.0 kg), MeCN (3.8 kg) and TEA (5.4 kg). The mixture was cooled to 5-10° C., and thionyl chloride (3.2 kg) was added slowly while cooling the solution to maintain the temperature below 20° C. The mixture was stirred until no starting material was left (GC analysis). The reaction was typically complete within 30 min after addition is complete. To the mixture was added water (9 kg) and after stirring, the mixture was allowed to settle. The aqueous phase was discarded and the organic phase was washed with a mixture of water (8 kg) and saturated NaHCO3 (4 kg) solution. To the remaining organic phase containing 36 was added MeCN (2.5 kg) and solid NaHCO3 (3.1 kg). The resulting slurry was cooled to ca. 10° C. Bleach (NaOCl solution, 6.89 wt % aqueous solution, 52.4 kg, 2 eq.) was added slowly while cooling to maintain temperature below 25° C. The mixture was aged with stirring over 90-120 min at 20-25° C., until the reaction was complete (GC analysis). After completion of the reaction, the mixture was cooled to ca. 10° C. and then quenched with aqueous Na2SO3 solution (15.1% w/w, 21 kg) while cooling to maintain temperature below 20° C. The quenched reaction mixture was filtered through a cartridge filter to remove inorganic solids. The filtrate was allowed to settle, and phases are separated and the aqueous phase is discarded. The organic layer was washed first with a mixture of water (11 kg) and saturated NaHCO3 solution (4.7 kg), then with of saturated NaHCO3 solution (5.1 kg). DIPEA (220 mL) was added to the organic phase and the resulting solution was filtered through CELITE® (bag filter) into a clean drum. The reactor was rinsed with isopropyl acetate (7 kg) and the rinse is transferred to the drum. The organic phase was then concentrated under vacuum (25-28 inches of Hg) while maintaining reactor jacket temperature at 45-50° C. to afford 26 as an oil (˜10 L). Additional DIPEA (280 mL) was added and the vacuum distillation was continued (jacket temperature 50-55° C.) until no more distillate was collected. (batch volume ca. 7 L).
- step 3—To the concentrated oil from step 2 containing 26 was added TEA (2.34 kg) and TEA-trihydrofluoride (1.63 kg). The mixture was heated to 85° C. for 2 h. The batch was sampled to monitor the progress of the reaction by GC. After the reaction was complete conc. HCl (2.35 kg) was added to the mixture and the resulting mixture heated to ca. 90° C. (small amount of distillate collected). The reaction mixture was stirred at ca. 90° C. for 30 min and then saturated aqueous BaCl2 solution (18.8 kg) was added. The resulting suspension was stirred at about 90° C. for 4 h. The resulting mixture was then azeotropically dried under a vacuum (9-10 inches of Hg) by adding slowly n-propanol (119 kg) while distilling off the azeotropic mixture (internal batch temperature ca. 85-90° C.). To the residual suspension was added toluene (33 kg) and vacuum distillation was continued to distill off residual n-propanol (and traces of water) to a minimum volume to afford 28.
- step 4—To the residue from step 3 containing 28 was added MeCN (35 kg) and ca. 15 L was distilled out under atmospheric pressure. The reaction mixture was cooled to ca. 10° C. and then benzoyl chloride (8.27 kg) and DMAP (0.14 kg) are added. TEA (5.84 kg) was added slowly to the reaction mixture while cooling to maintain temperature below 40° C. The batch was aged at ca. 20° C. and the progress of the benzoylation is monitored by HPLC. After completion of the reaction, EtOAc (30 kg) was added to the mixture and the resulting suspension is stirred for about 30 min. The reaction mixture was filtered through a CELITE® pad (using a nutsche filter) to remove inorganic salts. The solid cake was washed with EtOAc (38 kg). The combined filtrate and washes were washed successively with water (38 kg), saturated NaHCO3 solution (40 kg) and saturated brine (44 kg). The organic phase was polish-filtered (through a cartridge filter) and concentrated under modest vacuum to minimum volume. IPA (77 kg) was added to the concentrate and ca. 25 L of distillate was collected under modest vacuum allowing the internal batch temperature to reach ca. 75° C. at the end of the distillation. The remaining solution was then cooled to ca. 5° C. over 5 h and optionally aged overnight. The precipitate was filtered and washed with of cold (ca. 5° C.) IPA (24 kg). The product was dried under vacuum at 60-70° C. to afford 6.63 kg (70.7% theory of 10 which was 98.2% pure by HPLC.
- The features disclosed in the foregoing description, or the following claims, expressed in their specific forms or in terms of a means for performing the disclosed function, or a method or process for attaining the disclosed result, as appropriate, may, separately, or in any combination of such features, be utilized for realizing the invention in diverse forms thereof.
- The foregoing invention has been described in some detail by way of illustration and example, for purposes of clarity and understanding. It will be obvious to one of skill in the art that changes and modifications may be practiced within the scope of the appended claims. Therefore, it is to be understood that the above description is intended to be illustrative and not restrictive. The scope of the invention should, therefore, be determined not with reference to the above description, but should instead be determined with reference to the following appended claims, along with the full scope of equivalents to which such claims are entitled.
- All patents, patent applications and publications cited in this application are hereby incorporated by reference in their entirety for all purposes to the same extent as if each individual patent, patent application or publication were so individually denoted.
Claims (5)
1. A process for the preparation of a nucleoside of formula I comprising the steps of:

(i) contacting an (aryl)alkanoic acid (2R,3R,4R)-2-(aryl)alkanoyloxymethyl-4-fluoro-4-methyl-5-oxo-tetrahydro-furan-3-yl ester (II; R=aryl or alkyl) with RED-AL/TFE (1:1) in DCM/PhMe (6:1) at a temperature of −10 to −5° C.;
(ii) contacting the mixture with a chlorinating agent selected from sulfuryl chloride, thionyl chloride or phosphorus oxychloride and tetrabutylammonium bromide to afford an
(aryl)alkanoic acid (2R,3R,4R)-2-(aryl)alkanoyloxy methyl-5-chloro-4-fluoro-4-methyl-tetrahydro-furan-3-yl ester (III; R=aryl or alkyl); and,
(iii) contacting III with N-(2-trimethylsilanyloxy-pyrimidin-4-yl)-benzamide, SnCl4 and PhCl to afford an (aryl)alkanoic acid (2R,3R,4R,5R)-3-(aryl)alkanoyloxy-5-(4-benzoylamino-2-oxo-2H-pyrimidin-1-yl)-4-fluoro-4-methyl-tetrahydro-furan-2-ylmethyl ester (I; R=aryl or alkyl).
2. A process according to claim 1 said process further comprising the step of hydrolysis of the protected nucleoside I to 4-amino-1-((2R,3R,4R,5R)-3-fluoro-4-hydroxy-5-hydroxymethyl-3-methyl-tetrahydro-furan-2-yl)-1H-pyrimidin-2-one (IV).

3. A process according to claim 2 wherein R is C6H5.
4. A process according to claim 3 said process further comprising the steps of:

(i) contacting (E)-3-((S)-2,2-dimethyl-[1,3]dioxolan-4-yl)-2-methyl-acrylic acid ethyl ester (V) with aqueous sodium permanganate, ethylene glycol, sodium bicarbonate and acetone at −20 to 0° C., then quenching the reaction with aqueous sodium bisulfite, partitioning the product between EtOAc and water and recovering (2S,3R)-3-((R)-2,2-dimethyl-[1,3]dioxolan-4-yl)-2,3-dihydroxy-2-methyl-propionic acid ethyl ester (VI) from the organic phase;

(ii) contacting a solution of (VI) and TEA in iso-propyl acetate and MeCN with thionyl chloride to afford (VII);

(iii) contacting a solution of (VII) in iso-propyl acetate and MeCN with NaHCO3 and aqueous NaOCl to afford (VIII);

(iv) contacting (VIII) with TEA and triethylamine-trihydrofluoride to afford (IX) followed by HCl, BaCl2 and water to afford (X);

(v) contacting a solution of (X) in MeCN with TEA, DMAP and benzoyl chloride to afford II (R═C6H5).
5. A process for the preparation of an (aryl)alkanoic acid (2R,3R,4R)-2-(aryl)alkanoyloxy methyl-5-chloro-4-fluoro-4-methyl-tetrahydro-furan-3-yl ester (III, R=aryl or alkyl) from an (aryl)alkanoic acid (2R,3R,4R)-2-(aryl)alkanoyloxymethyl-4-fluoro-4-methyl-5-oxo-tetrahydro-furan-3-yl ester (II, R=aryl or alkyl) comprising the steps of:

(i) contacting II with RED-AL/TFE (1:1) in DCM/PhMe (6:1) at a temperature of −10 to −5° C.; and, subsequently
(ii) treating the mixture with sulfuryl chloride and tetrabutylammonium bromide to afford (III).
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US11/973,748 US20080139802A1 (en) | 2006-10-10 | 2007-10-10 | Preparation of nucleosides ribofuranosyl pyrimidines |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US85096206P | 2006-10-10 | 2006-10-10 | |
| US11/973,748 US20080139802A1 (en) | 2006-10-10 | 2007-10-10 | Preparation of nucleosides ribofuranosyl pyrimidines |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| US20080139802A1 true US20080139802A1 (en) | 2008-06-12 |
Family
ID=39111500
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US12/444,608 Active 2031-04-23 US8912321B2 (en) | 2006-10-10 | 2007-10-05 | Preparation of nucleosides ribofuranosyl pyrimidines |
| US11/973,748 Abandoned US20080139802A1 (en) | 2006-10-10 | 2007-10-10 | Preparation of nucleosides ribofuranosyl pyrimidines |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US12/444,608 Active 2031-04-23 US8912321B2 (en) | 2006-10-10 | 2007-10-05 | Preparation of nucleosides ribofuranosyl pyrimidines |
Country Status (19)
| Country | Link |
|---|---|
| US (2) | US8912321B2 (en) |
| EP (1) | EP2084174B1 (en) |
| JP (1) | JP5252459B2 (en) |
| KR (1) | KR101057239B1 (en) |
| CN (1) | CN101600725B (en) |
| AU (1) | AU2007307057B2 (en) |
| BR (1) | BRPI0719174A2 (en) |
| CA (1) | CA2666098C (en) |
| DK (1) | DK2084174T3 (en) |
| ES (1) | ES2429290T3 (en) |
| GT (1) | GT200900080A (en) |
| HK (1) | HK1133889A1 (en) |
| IL (1) | IL198086A (en) |
| MX (1) | MX2009003795A (en) |
| PL (1) | PL2084174T3 (en) |
| PT (1) | PT2084174E (en) |
| RU (1) | RU2421461C2 (en) |
| SI (1) | SI2084174T1 (en) |
| WO (1) | WO2008045419A1 (en) |
Cited By (32)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2010075517A2 (en) | 2008-12-23 | 2010-07-01 | Pharmasset, Inc. | Nucleoside analogs |
| WO2010075549A2 (en) | 2008-12-23 | 2010-07-01 | Pharmasset, Inc. | Nucleoside phosphoramidates |
| WO2010075554A1 (en) | 2008-12-23 | 2010-07-01 | Pharmasset, Inc. | Synthesis of purine nucleosides |
| WO2011123668A2 (en) | 2010-03-31 | 2011-10-06 | Pharmasset, Inc. | Stereoselective synthesis of phosphorus containing actives |
| US8173621B2 (en) | 2008-06-11 | 2012-05-08 | Gilead Pharmasset Llc | Nucleoside cyclicphosphates |
| US8507460B2 (en) | 2011-10-14 | 2013-08-13 | Idenix Pharmaceuticals, Inc. | Substituted 3′,5′-cyclic phosphates of purine nucleotide compounds and pharmaceutical compositions for the treatment of viral infections |
| US8563530B2 (en) | 2010-03-31 | 2013-10-22 | Gilead Pharmassel LLC | Purine nucleoside phosphoramidate |
| US8669382B2 (en) | 2010-06-03 | 2014-03-11 | Central Glass Company, Limited | Method for producing (2R)-2-fluoro-2-C-methyl-D-ribono-γ-lactone precursor |
| US8841275B2 (en) | 2010-11-30 | 2014-09-23 | Gilead Pharmasset Llc | 2′-spiro-nucleosides and derivatives thereof useful for treating hepatitis C virus and dengue virus infections |
| US9109001B2 (en) | 2012-05-22 | 2015-08-18 | Idenix Pharmaceuticals, Inc. | 3′,5′-cyclic phosphoramidate prodrugs for HCV infection |
| US9187515B2 (en) | 2013-04-01 | 2015-11-17 | Idenix Pharmaceuticals Llc | 2′,4′-fluoro nucleosides for the treatment of HCV |
| US9192621B2 (en) | 2012-09-27 | 2015-11-24 | Idenix Pharmaceuticals Llc | Esters and malonates of SATE prodrugs |
| US9211300B2 (en) | 2012-12-19 | 2015-12-15 | Idenix Pharmaceuticals Llc | 4′-fluoro nucleosides for the treatment of HCV |
| US9243025B2 (en) | 2011-03-31 | 2016-01-26 | Idenix Pharmaceuticals, Llc | Compounds and pharmaceutical compositions for the treatment of viral infections |
| US9296778B2 (en) | 2012-05-22 | 2016-03-29 | Idenix Pharmaceuticals, Inc. | 3′,5′-cyclic phosphate prodrugs for HCV infection |
| US9309275B2 (en) | 2013-03-04 | 2016-04-12 | Idenix Pharmaceuticals Llc | 3′-deoxy nucleosides for the treatment of HCV |
| US9339541B2 (en) | 2013-03-04 | 2016-05-17 | Merck Sharp & Dohme Corp. | Thiophosphate nucleosides for the treatment of HCV |
| US9403863B2 (en) | 2011-09-12 | 2016-08-02 | Idenix Pharmaceuticals Llc | Substituted carbonyloxymethylphosphoramidate compounds and pharmaceutical compositions for the treatment of viral infections |
| US9422323B2 (en) | 2012-05-25 | 2016-08-23 | Janssen Sciences Ireland Uc | Uracyl spirooxetane nucleosides |
| US9675632B2 (en) | 2014-08-26 | 2017-06-13 | Enanta Pharmaceuticals, Inc. | Nucleoside and nucleotide derivatives |
| US9718851B2 (en) | 2014-11-06 | 2017-08-01 | Enanta Pharmaceuticals, Inc. | Deuterated nucleoside/tide derivatives |
| US9732110B2 (en) | 2014-12-05 | 2017-08-15 | Enanta Pharmaceuticals, Inc. | Nucleoside and nucleotide derivatives |
| US10005779B2 (en) | 2013-06-05 | 2018-06-26 | Idenix Pharmaceuticals Llc | 1′,4′-thio nucleosides for the treatment of HCV |
| US10034893B2 (en) | 2013-02-01 | 2018-07-31 | Enanta Pharmaceuticals, Inc. | 5, 6-D2 uridine nucleoside/tide derivatives |
| US10202411B2 (en) | 2014-04-16 | 2019-02-12 | Idenix Pharmaceuticals Llc | 3′-substituted methyl or alkynyl nucleosides nucleotides for the treatment of HCV |
| US10231986B2 (en) | 2013-03-13 | 2019-03-19 | Idenix Pharmaceuticals Llc | Amino acid phosphoramidate pronucleotides of 2′-cyano, azido and amino nucleosides for the treatment of HCV |
| US10238680B2 (en) | 2013-08-01 | 2019-03-26 | Idenix Pharmaceuticals Llc | D-amino acid phosphoramidate pronucleotides of halogeno pyrimidine compounds for liver disease |
| US10363265B2 (en) | 2000-05-23 | 2019-07-30 | Idenix Pharmaceuticals Llc | Methods and compositions for treating hepatitis C virus |
| US10513534B2 (en) | 2012-10-08 | 2019-12-24 | Idenix Pharmaceuticals Llc | 2′-chloro nucleoside analogs for HCV infection |
| US10525072B2 (en) | 2002-11-15 | 2020-01-07 | Idenix Pharmaceuticals Llc | 2′-branched nucleosides and flaviviridae mutation |
| US10717758B2 (en) | 2012-05-22 | 2020-07-21 | Idenix Pharmaceuticals Llc | D-amino acid compounds for liver disease |
| US10723754B2 (en) | 2012-10-22 | 2020-07-28 | Idenix Pharmaceuticals Llc | 2′,4′-bridged nucleosides for HCV infection |
Families Citing this family (47)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA2734052A1 (en) | 2003-05-30 | 2005-01-13 | Pharmasset, Inc. | Modified fluorinated nucleoside analogues |
| PL3109244T3 (en) * | 2004-09-14 | 2019-09-30 | Gilead Pharmasset Llc | Preparation of 2'fluoro-2'-alkyl-substituted or other optionally substituted ribofuranosyl pyrimidines and purines and their derivatives |
| CA2587118C (en) | 2004-11-16 | 2014-12-30 | Thomson Licensing | Film grain sei message insertion for bit-accurate simulation in a video system |
| AU2005306921B2 (en) | 2004-11-16 | 2011-03-03 | Interdigital Vc Holdings, Inc. | Film grain simulation method based on pre-computed transform coefficients |
| HUE044545T2 (en) | 2004-11-17 | 2019-10-28 | Interdigital Vc Holdings Inc | Bit-accurate film grain simulation method based on pre-computed transformed coefficients |
| US7964580B2 (en) | 2007-03-30 | 2011-06-21 | Pharmasset, Inc. | Nucleoside phosphoramidate prodrugs |
| US10715834B2 (en) | 2007-05-10 | 2020-07-14 | Interdigital Vc Holdings, Inc. | Film grain simulation based on pre-computed transform coefficients |
| US8618076B2 (en) | 2009-05-20 | 2013-12-31 | Gilead Pharmasset Llc | Nucleoside phosphoramidates |
| TWI576352B (en) | 2009-05-20 | 2017-04-01 | 基利法瑪席特有限責任公司 | Nucleoside phosphoramidates |
| WO2011123672A1 (en) | 2010-03-31 | 2011-10-06 | Pharmasset, Inc. | Purine nucleoside phosphoramidate |
| JP6073897B2 (en) | 2011-09-16 | 2017-02-01 | ギリアド ファーマセット エルエルシー | Methods for treating HCV |
| US8466159B2 (en) | 2011-10-21 | 2013-06-18 | Abbvie Inc. | Methods for treating HCV |
| EP2583680A3 (en) | 2011-10-21 | 2013-06-12 | Abbvie Inc. | Mono (PSI-7977) or combination treatment of DAAs for use in treating HCV |
| US8492386B2 (en) | 2011-10-21 | 2013-07-23 | Abbvie Inc. | Methods for treating HCV |
| ES2572328B1 (en) | 2011-10-21 | 2017-08-24 | Abbvie Inc. | COMBINATION OF AT LEAST TWO ANTIVIRAL AGENTS OF DIRECT ACTION AND RIBAVIRINA BUT NOT INTERFERED, FOR USE IN THE TREATMENT OF HCV |
| US8889159B2 (en) | 2011-11-29 | 2014-11-18 | Gilead Pharmasset Llc | Compositions and methods for treating hepatitis C virus |
| MX2014014087A (en) | 2012-05-29 | 2015-01-26 | Hoffmann La Roche | Process for the preparation of 2-deoxy-2-fluoro-2-methyl-d-ribofu ranosyl nucleoside compounds. |
| CN103450148B (en) * | 2012-06-04 | 2018-06-12 | 浙江九洲药业股份有限公司 | A kind of synthetic method of five-membered ring sulfate compound |
| CN103724301A (en) | 2012-10-10 | 2014-04-16 | 上海特化医药科技有限公司 | (2R)-2-desoxy-2,2-disubstituted-1,4-ribonolactones, preparation method and purpose thereof |
| CN103012386B (en) * | 2012-12-26 | 2015-04-01 | 浙江普洛得邦制药有限公司 | Preparation method of five-membered cyclic sulphate |
| BR112014011938B1 (en) | 2013-01-31 | 2021-03-16 | Gilead Pharmasset Llc | pharmaceutical composition in the form of a tablet with a fixed dose combination of two antiviral compounds, a pharmaceutical dosage form comprising said composition and use of said composition |
| EA201690473A1 (en) | 2013-08-27 | 2017-03-31 | ГАЙЛИД ФАРМАССЕТ ЭлЭлСи | COMBINED COMPOSITION OF TWO ANTI-VIRUS COMPOUNDS |
| CN104447923B (en) * | 2013-09-23 | 2018-03-30 | 中国药科大学 | Methyl nucleoside derivatives of 2 ' deoxidation, 2 ' fluorine 2 ' and preparation method thereof and the purposes in pharmacy |
| WO2016042576A1 (en) | 2014-09-16 | 2016-03-24 | Cadila Healthcare Limited | Co-crystal of sofosbuvir and amino acid and process for preparation thereof |
| CN104744539A (en) * | 2014-09-19 | 2015-07-01 | 上海皓元生物医药科技有限公司 | Synthesis method for (2'R)-2'-deoxy-2'-fluorine-2'-methyl uridine |
| CN104327138B (en) * | 2014-10-21 | 2017-05-10 | 齐鲁制药有限公司 | Preparation method of PSI-7977 intermediate compound |
| CN104610404B (en) * | 2015-01-16 | 2016-04-06 | 南通常佑药业科技有限公司 | A kind of preparation method of ribofuranose phosphate derivative |
| SG11201706841PA (en) | 2015-03-06 | 2017-09-28 | Atea Pharmaceuticals Inc | ß-D-2'-DEOXY-2'a-FLUORO-2'-ß-C-SUBSTITUTED-2-MODIFIED-N<sp>6</sp>-SUBSTITUTED PURINE NUCLEOTIDES FOR HCV TREATMENT |
| CN104829672B (en) * | 2015-05-19 | 2018-03-13 | 江苏福瑞生物医药有限公司 | A kind of synthetic method of pharmaceutical intermediate |
| CN105061535A (en) * | 2015-09-02 | 2015-11-18 | 江苏科本医药化学有限公司 | Synthetic method of sofosbuvir intermediate |
| CN106554333B (en) * | 2015-09-29 | 2018-11-30 | 江苏福瑞生物医药有限公司 | A kind of synthetic method of pharmaceutical intermediate |
| CN106608896B (en) * | 2015-10-26 | 2019-08-27 | 江苏福瑞康泰药业有限公司 | A kind of synthetic method of pharmaceutical intermediate |
| CN105418547A (en) * | 2015-11-17 | 2016-03-23 | 海门慧聚药业有限公司 | Preparation for sofosbuvir key intermediate |
| CN105503983B (en) * | 2015-12-17 | 2019-06-28 | 江苏阿尔法药业有限公司 | The preparation method of Suo Feibuwei intermediate and its derivative |
| CN105566422B (en) * | 2015-12-29 | 2019-06-25 | 江苏阿尔法药业有限公司 | The preparation method of Suo Feibuwei intermediate or derivatives thereof |
| EP3448392A4 (en) | 2016-04-28 | 2020-01-15 | Emory University | Alkyne containing nucleotide and nucleoside therapeutic compositions and uses related thereto |
| US10711029B2 (en) | 2016-07-14 | 2020-07-14 | Atea Pharmaceuticals, Inc. | Beta-d-2′-deoxy-2′-alpha-fluoro-2′-beta-c-substituted-4′fluoro-n6-substituted-6-amino-2-substituted purine nucleotides for the treatment of hepatitis c virus infection |
| US10239910B2 (en) | 2016-07-20 | 2019-03-26 | Optimus Drugs (P) Limited | Process for the preparation of sofosbuvir |
| MY197236A (en) | 2016-09-07 | 2023-06-07 | Atea Pharmaceuticals Inc | 2'-substituted-n6-substituted purine nucleotides for rna virus treatment |
| KR102592899B1 (en) | 2017-02-01 | 2023-10-24 | 아테아 파마슈티컬즈, 인크. | Nucleotide hemi-sulfate salt for the treatment of hepatitis c virus |
| CN106810515A (en) * | 2017-02-06 | 2017-06-09 | 抚州市星辰药业有限公司 | The midbody compound and its synthetic method of a kind of synthesis Suo Feibuwei |
| CN109422789A (en) * | 2017-08-28 | 2019-03-05 | 常州制药厂有限公司 | A kind of preparation process amelioration method of Suo Feibuwei |
| CN112351799A (en) | 2018-04-10 | 2021-02-09 | 阿堤亚制药公司 | Treatment of HCV infected patients with cirrhosis |
| CN108675984A (en) * | 2018-06-22 | 2018-10-19 | 江苏阿尔法药业有限公司 | The preparation method of Suo Feibuwei intermediates |
| CN111606961A (en) * | 2019-02-26 | 2020-09-01 | 顾世海 | Process production method of (2 'R) -2' -deoxy-2 '-fluoro-2' -methyluridine |
| CN111040010A (en) * | 2019-12-23 | 2020-04-21 | 上海红蓝医药科技有限公司 | Synthetic method of sofosbuvir intermediate |
| US10874687B1 (en) | 2020-02-27 | 2020-12-29 | Atea Pharmaceuticals, Inc. | Highly active compounds against COVID-19 |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20040077587A1 (en) * | 2002-06-28 | 2004-04-22 | Jean-Pierre Sommadossi | 2'-C-methyl-3'-O-L-valine ester ribofuranosyl cytidine for treatment of flaviviridae infections |
| US20050009737A1 (en) * | 2003-05-30 | 2005-01-13 | Jeremy Clark | Modified fluorinated nucleoside analogues |
| US20060122146A1 (en) * | 2004-09-14 | 2006-06-08 | Byoung-Kwon Chun | Preparation of 2'-fluoro-2'-alkyl-substituted or other optionally substituted ribofuranosyl pyrimidines and purines and their derivatives |
Family Cites Families (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR850002973A (en) | 1983-10-03 | 1985-05-28 | 구라바야시 이꾸시로 | Method for preparing cytidine derivative |
| BR9908270A (en) | 1998-02-25 | 2004-06-29 | Univ Emory | 2-Fluoro-nucleosides, pharmaceutical compositions and their uses |
| KR20020091829A (en) | 1999-07-30 | 2002-12-06 | 애보트 게엠베하 운트 콤파니 카게 | 2-Pyrazolin-5-ones |
| MY164523A (en) | 2000-05-23 | 2017-12-29 | Univ Degli Studi Cagliari | Methods and compositions for treating hepatitis c virus |
| CZ301182B6 (en) | 2000-05-26 | 2009-12-02 | Idenix (Cayman) Limited | Use of nucleoside derivatives for preparation of pharmaceutical compositions for treating infections caused by flaviviruses and pestiviruses |
| ATE526339T1 (en) | 2001-01-22 | 2011-10-15 | Merck Sharp & Dohme | NUCLEOSIDE DERIVATIVES AS INHIBITORS OF RNA-DEPENDENT VIRAL RNA POLYMERASE |
| US7105499B2 (en) | 2001-01-22 | 2006-09-12 | Merck & Co., Inc. | Nucleoside derivatives as inhibitors of RNA-dependent RNA viral polymerase |
| EP1435974A4 (en) | 2001-09-28 | 2006-09-06 | Idenix Cayman Ltd | Methods and compositions for treating hepatitis c virus using 4'-modified nucleosides |
| US7608600B2 (en) | 2002-06-28 | 2009-10-27 | Idenix Pharmaceuticals, Inc. | Modified 2′ and 3′-nucleoside prodrugs for treating Flaviviridae infections |
| EP2799442A1 (en) | 2002-06-28 | 2014-11-05 | IDENIX Pharmaceuticals, Inc. | Modified 2' and 3' -nucleoside prodrugs for treating flaviridae infections |
| CA2506129C (en) | 2002-11-15 | 2015-02-17 | Idenix (Cayman) Limited | 2'-branched nucleosides and flaviviridae mutation |
| TWI332507B (en) | 2002-11-19 | 2010-11-01 | Hoffmann La Roche | Antiviral nucleoside derivatives |
| AU2004258750A1 (en) | 2003-07-25 | 2005-02-03 | Centre National De La Recherche Scientifique -Cnrs | Purine nucleoside analogues for treating diseases caused by flaviviridae including hepatitis C |
| RU2006109491A (en) | 2003-08-27 | 2006-08-10 | Байота, Инк. (Au) | NEW TRICYCLIC NUCLEOSIDES OR NUCLEOTIDES AS A THERAPEUTIC MEDICINES |
| PT1741716E (en) * | 2004-04-30 | 2011-12-21 | Daiichi Sankyo Co Ltd | Process for producing pentacyclic taxane |
| AU2005267051B2 (en) * | 2004-07-21 | 2011-07-14 | Gilead Pharmasset Llc | Preparation of alkyl-substituted 2-deoxy-2-fluoro-D-ribofuranosyl pyrimidines and purines and their derivatives |
-
2007
- 2007-10-05 RU RU2009117396/04A patent/RU2421461C2/en not_active IP Right Cessation
- 2007-10-05 US US12/444,608 patent/US8912321B2/en active Active
- 2007-10-05 MX MX2009003795A patent/MX2009003795A/en active IP Right Grant
- 2007-10-05 ES ES07839369T patent/ES2429290T3/en active Active
- 2007-10-05 DK DK07839369.1T patent/DK2084174T3/en active
- 2007-10-05 JP JP2009532372A patent/JP5252459B2/en active Active
- 2007-10-05 PL PL07839369T patent/PL2084174T3/en unknown
- 2007-10-05 CN CN200780037855.XA patent/CN101600725B/en active Active
- 2007-10-05 PT PT78393691T patent/PT2084174E/en unknown
- 2007-10-05 KR KR1020097008991A patent/KR101057239B1/en active IP Right Grant
- 2007-10-05 WO PCT/US2007/021548 patent/WO2008045419A1/en active Application Filing
- 2007-10-05 SI SI200731311T patent/SI2084174T1/en unknown
- 2007-10-05 AU AU2007307057A patent/AU2007307057B2/en active Active
- 2007-10-05 CA CA2666098A patent/CA2666098C/en active Active
- 2007-10-05 EP EP07839369.1A patent/EP2084174B1/en active Active
- 2007-10-05 BR BRPI0719174A patent/BRPI0719174A2/en not_active IP Right Cessation
- 2007-10-10 US US11/973,748 patent/US20080139802A1/en not_active Abandoned
-
2009
- 2009-04-07 IL IL198086A patent/IL198086A/en active IP Right Grant
- 2009-04-13 GT GT200900080A patent/GT200900080A/en unknown
-
2010
- 2010-02-02 HK HK10101126.3A patent/HK1133889A1/en unknown
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20040077587A1 (en) * | 2002-06-28 | 2004-04-22 | Jean-Pierre Sommadossi | 2'-C-methyl-3'-O-L-valine ester ribofuranosyl cytidine for treatment of flaviviridae infections |
| US20050009737A1 (en) * | 2003-05-30 | 2005-01-13 | Jeremy Clark | Modified fluorinated nucleoside analogues |
| US20060122146A1 (en) * | 2004-09-14 | 2006-06-08 | Byoung-Kwon Chun | Preparation of 2'-fluoro-2'-alkyl-substituted or other optionally substituted ribofuranosyl pyrimidines and purines and their derivatives |
Cited By (49)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10363265B2 (en) | 2000-05-23 | 2019-07-30 | Idenix Pharmaceuticals Llc | Methods and compositions for treating hepatitis C virus |
| US10758557B2 (en) | 2000-05-23 | 2020-09-01 | Idenix Pharmaceuticals Llc | Methods and compositions for treating hepatitis C virus |
| US10525072B2 (en) | 2002-11-15 | 2020-01-07 | Idenix Pharmaceuticals Llc | 2′-branched nucleosides and flaviviridae mutation |
| US8759510B2 (en) | 2008-06-11 | 2014-06-24 | Gilead Pharmasset Llc | Nucleoside cyclicphosphates |
| US8173621B2 (en) | 2008-06-11 | 2012-05-08 | Gilead Pharmasset Llc | Nucleoside cyclicphosphates |
| WO2010075517A2 (en) | 2008-12-23 | 2010-07-01 | Pharmasset, Inc. | Nucleoside analogs |
| US8551973B2 (en) | 2008-12-23 | 2013-10-08 | Gilead Pharmasset Llc | Nucleoside analogs |
| EP3222628A1 (en) | 2008-12-23 | 2017-09-27 | Gilead Pharmasset LLC | Nucleoside phosphoramidates |
| EP2671888A1 (en) | 2008-12-23 | 2013-12-11 | Gilead Pharmasset LLC | 3',5'-cyclic nucleoside phosphate analogues |
| US8957045B2 (en) | 2008-12-23 | 2015-02-17 | Gilead Pharmasset Llc | Nucleoside phosphoramidates |
| US8716262B2 (en) | 2008-12-23 | 2014-05-06 | Gilead Pharmasset Llc | Nucleoside phosphoramidates |
| US8716263B2 (en) | 2008-12-23 | 2014-05-06 | Gilead Pharmasset Llc | Synthesis of purine nucleosides |
| WO2010075554A1 (en) | 2008-12-23 | 2010-07-01 | Pharmasset, Inc. | Synthesis of purine nucleosides |
| WO2010075549A2 (en) | 2008-12-23 | 2010-07-01 | Pharmasset, Inc. | Nucleoside phosphoramidates |
| US9045520B2 (en) | 2008-12-23 | 2015-06-02 | Gilead Pharmasset Llc | Synthesis of purine nucleosides |
| US8563530B2 (en) | 2010-03-31 | 2013-10-22 | Gilead Pharmassel LLC | Purine nucleoside phosphoramidate |
| US8859756B2 (en) | 2010-03-31 | 2014-10-14 | Gilead Pharmasset Llc | Stereoselective synthesis of phosphorus containing actives |
| EP2752422A1 (en) | 2010-03-31 | 2014-07-09 | Gilead Pharmasset LLC | Stereoselective synthesis of phosphorus containing actives |
| WO2011123668A2 (en) | 2010-03-31 | 2011-10-06 | Pharmasset, Inc. | Stereoselective synthesis of phosphorus containing actives |
| US8669382B2 (en) | 2010-06-03 | 2014-03-11 | Central Glass Company, Limited | Method for producing (2R)-2-fluoro-2-C-methyl-D-ribono-γ-lactone precursor |
| US8841275B2 (en) | 2010-11-30 | 2014-09-23 | Gilead Pharmasset Llc | 2′-spiro-nucleosides and derivatives thereof useful for treating hepatitis C virus and dengue virus infections |
| US9394331B2 (en) | 2010-11-30 | 2016-07-19 | Gilead Pharmasset Llc | 2′-spiro-nucleosides and derivatives thereof useful for treating hepatitis C virus and dengue virus infections |
| US9243025B2 (en) | 2011-03-31 | 2016-01-26 | Idenix Pharmaceuticals, Llc | Compounds and pharmaceutical compositions for the treatment of viral infections |
| US9403863B2 (en) | 2011-09-12 | 2016-08-02 | Idenix Pharmaceuticals Llc | Substituted carbonyloxymethylphosphoramidate compounds and pharmaceutical compositions for the treatment of viral infections |
| US8507460B2 (en) | 2011-10-14 | 2013-08-13 | Idenix Pharmaceuticals, Inc. | Substituted 3′,5′-cyclic phosphates of purine nucleotide compounds and pharmaceutical compositions for the treatment of viral infections |
| US9109001B2 (en) | 2012-05-22 | 2015-08-18 | Idenix Pharmaceuticals, Inc. | 3′,5′-cyclic phosphoramidate prodrugs for HCV infection |
| US10717758B2 (en) | 2012-05-22 | 2020-07-21 | Idenix Pharmaceuticals Llc | D-amino acid compounds for liver disease |
| US9296778B2 (en) | 2012-05-22 | 2016-03-29 | Idenix Pharmaceuticals, Inc. | 3′,5′-cyclic phosphate prodrugs for HCV infection |
| US10040814B2 (en) | 2012-05-25 | 2018-08-07 | Janssen Sciences Ireland Uc | Uracyl spirooxetane nucleosides |
| US10544184B2 (en) | 2012-05-25 | 2020-01-28 | Janssen Sciences Ireland Unlimited Company | Uracyl spirooxetane nucleosides |
| US10774106B2 (en) | 2012-05-25 | 2020-09-15 | Janssen Sciences Ireland Unlimited Company | Uracyl spirooxetane nucleosides |
| US9422323B2 (en) | 2012-05-25 | 2016-08-23 | Janssen Sciences Ireland Uc | Uracyl spirooxetane nucleosides |
| US9845336B2 (en) | 2012-05-25 | 2017-12-19 | Janssen Sciences Ireland Uc | Uracyl spirooxetane nucleosides |
| US10301347B2 (en) | 2012-05-25 | 2019-05-28 | Janssen Sciences Ireland Unlimited Company | Uracyl spirooxetane nucleosides |
| US9192621B2 (en) | 2012-09-27 | 2015-11-24 | Idenix Pharmaceuticals Llc | Esters and malonates of SATE prodrugs |
| US10513534B2 (en) | 2012-10-08 | 2019-12-24 | Idenix Pharmaceuticals Llc | 2′-chloro nucleoside analogs for HCV infection |
| US10723754B2 (en) | 2012-10-22 | 2020-07-28 | Idenix Pharmaceuticals Llc | 2′,4′-bridged nucleosides for HCV infection |
| US9211300B2 (en) | 2012-12-19 | 2015-12-15 | Idenix Pharmaceuticals Llc | 4′-fluoro nucleosides for the treatment of HCV |
| US10034893B2 (en) | 2013-02-01 | 2018-07-31 | Enanta Pharmaceuticals, Inc. | 5, 6-D2 uridine nucleoside/tide derivatives |
| US9339541B2 (en) | 2013-03-04 | 2016-05-17 | Merck Sharp & Dohme Corp. | Thiophosphate nucleosides for the treatment of HCV |
| US9309275B2 (en) | 2013-03-04 | 2016-04-12 | Idenix Pharmaceuticals Llc | 3′-deoxy nucleosides for the treatment of HCV |
| US10231986B2 (en) | 2013-03-13 | 2019-03-19 | Idenix Pharmaceuticals Llc | Amino acid phosphoramidate pronucleotides of 2′-cyano, azido and amino nucleosides for the treatment of HCV |
| US9187515B2 (en) | 2013-04-01 | 2015-11-17 | Idenix Pharmaceuticals Llc | 2′,4′-fluoro nucleosides for the treatment of HCV |
| US10005779B2 (en) | 2013-06-05 | 2018-06-26 | Idenix Pharmaceuticals Llc | 1′,4′-thio nucleosides for the treatment of HCV |
| US10238680B2 (en) | 2013-08-01 | 2019-03-26 | Idenix Pharmaceuticals Llc | D-amino acid phosphoramidate pronucleotides of halogeno pyrimidine compounds for liver disease |
| US10202411B2 (en) | 2014-04-16 | 2019-02-12 | Idenix Pharmaceuticals Llc | 3′-substituted methyl or alkynyl nucleosides nucleotides for the treatment of HCV |
| US9675632B2 (en) | 2014-08-26 | 2017-06-13 | Enanta Pharmaceuticals, Inc. | Nucleoside and nucleotide derivatives |
| US9718851B2 (en) | 2014-11-06 | 2017-08-01 | Enanta Pharmaceuticals, Inc. | Deuterated nucleoside/tide derivatives |
| US9732110B2 (en) | 2014-12-05 | 2017-08-15 | Enanta Pharmaceuticals, Inc. | Nucleoside and nucleotide derivatives |
Also Published As
| Publication number | Publication date |
|---|---|
| IL198086A0 (en) | 2009-12-24 |
| KR20090060460A (en) | 2009-06-12 |
| CN101600725B (en) | 2014-11-26 |
| RU2009117396A (en) | 2010-11-20 |
| AU2007307057B2 (en) | 2011-09-15 |
| CN101600725A (en) | 2009-12-09 |
| CA2666098A1 (en) | 2008-04-17 |
| KR101057239B1 (en) | 2011-08-16 |
| JP5252459B2 (en) | 2013-07-31 |
| RU2421461C2 (en) | 2011-06-20 |
| AU2007307057A1 (en) | 2008-04-17 |
| JP2010505954A (en) | 2010-02-25 |
| DK2084174T3 (en) | 2013-11-04 |
| WO2008045419A1 (en) | 2008-04-17 |
| EP2084174B1 (en) | 2013-07-31 |
| PL2084174T3 (en) | 2013-12-31 |
| IL198086A (en) | 2012-03-29 |
| US20100056770A1 (en) | 2010-03-04 |
| GT200900080A (en) | 2012-04-10 |
| ES2429290T3 (en) | 2013-11-14 |
| BRPI0719174A2 (en) | 2017-06-13 |
| CA2666098C (en) | 2012-09-25 |
| EP2084174A1 (en) | 2009-08-05 |
| PT2084174E (en) | 2013-10-08 |
| HK1133889A1 (en) | 2010-04-09 |
| SI2084174T1 (en) | 2013-10-30 |
| US8912321B2 (en) | 2014-12-16 |
| MX2009003795A (en) | 2009-06-18 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US20080139802A1 (en) | Preparation of nucleosides ribofuranosyl pyrimidines | |
| US6639059B1 (en) | Synthesis of [2.2.1]bicyclo nucleosides | |
| US5401838A (en) | Stereoselective fusion glycosylation process for preparing 2'-deoxy-2',2'-difluoronucleosides and 2'-deoxy-2'-fluoronucleosides | |
| US20030092905A1 (en) | Synthesis of [2.2.1]bicyclo nucleosides | |
| KR20060026426A (en) | Processes for preparing 4'-azido nucleoside derivatives | |
| CN100335492C (en) | Process for the production of 3'-nucleoside prodrugs | |
| US8193354B2 (en) | Process for preparation of Gemcitabine hydrochloride | |
| US7485716B2 (en) | Stereoselective synthesis of β-nucleosides | |
| JP5766703B2 (en) | Synthesis of decitabine | |
| EP0638586B1 (en) | Nucleoside derivatives and methods for producing them | |
| EP1812457A1 (en) | STEREOSELECTIVE SYNTHESIS OF ß-NUCLEOSIDES | |
| US7439344B2 (en) | Selective O-acylation of nucleosides | |
| EP0640614A2 (en) | Stereoselective process for preparing beta-anomer enriched 2-deoxy-2, 2-difluoro-d-ribofuranosyl-3, 5-hydroxy protected-1-alkyl and aryl sulfonate intermediates | |
| EP0653437B1 (en) | Process for preparing AZT and derivatives thereof | |
| AU2009313842B2 (en) | Method of preparing deoxyribofuranose compounds | |
| WO1999038879A1 (en) | Process for producing nucleoside derivatives | |
| JPH08134065A (en) | Production of 3'-amino-3'-deoxynucleoside and intermediate for synthesizing the same | |
| JPH09110893A (en) | Production of 3'-amino-3'-deoxynucleoside |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AS | Assignment |
Owner name: PHARMASSET, INC, NEW JERSEY Free format text: ASSIGNMENT OF ASSIGNORS INTEREST;ASSIGNORS:CHUN, BYOUNG-KWON;RACHAKONDA, SUGUNA;ROSS, BRUCE;REEL/FRAME:020490/0735;SIGNING DATES FROM 20071116 TO 20071119 Owner name: ROCHE PALO ALTO LLC, CALIFORNIA Free format text: ASSIGNMENT OF ASSIGNORS INTEREST;ASSIGNORS:AXT, STEVEN D.;JIN, QINGWU;SARMA, KESHAB;AND OTHERS;REEL/FRAME:020490/0678 Effective date: 20071109 |
|
| STCB | Information on status: application discontinuation |
Free format text: ABANDONED -- FAILURE TO RESPOND TO AN OFFICE ACTION |