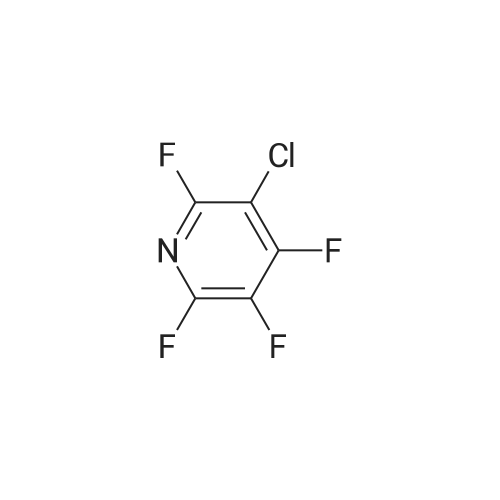
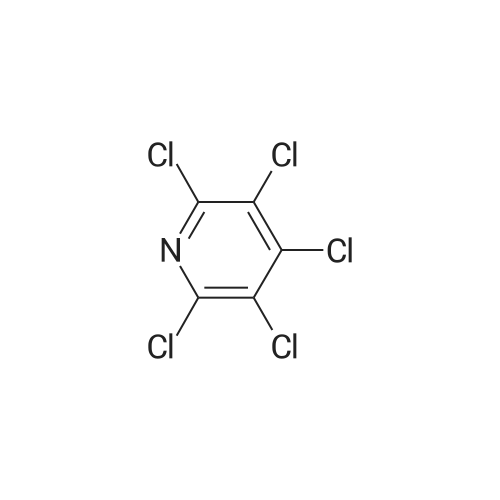
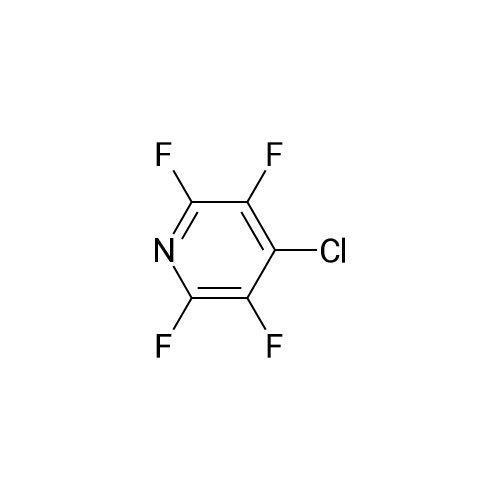
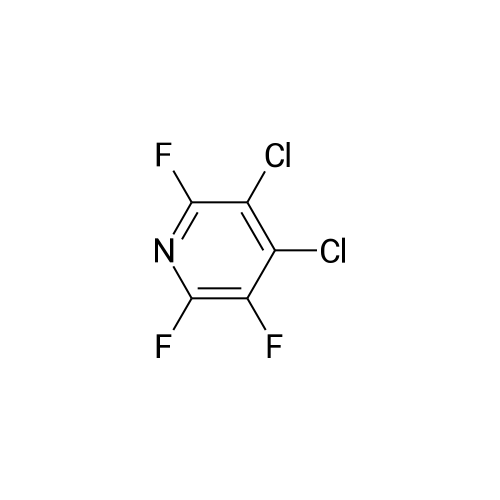
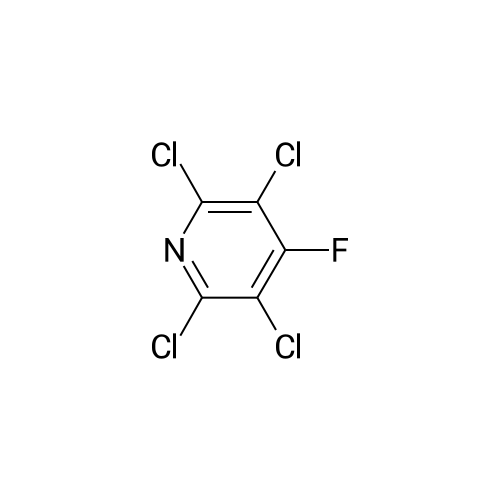
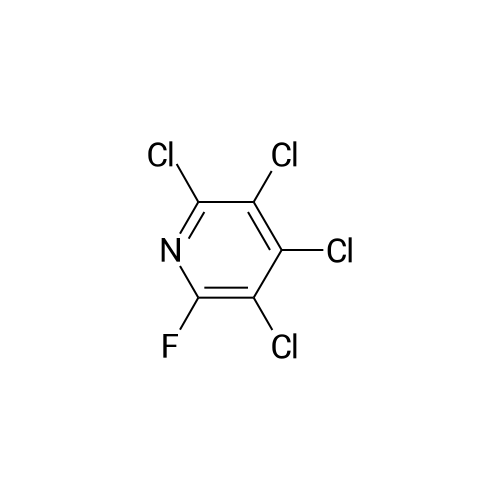
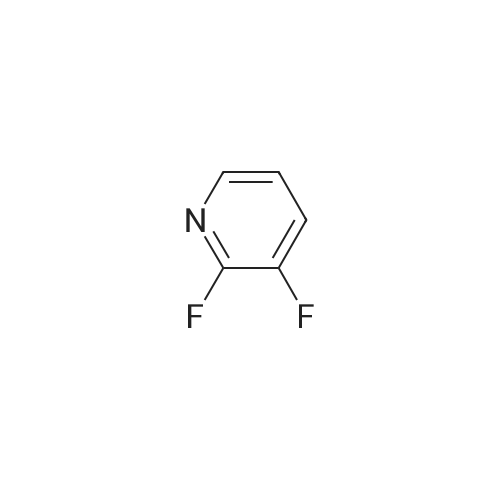
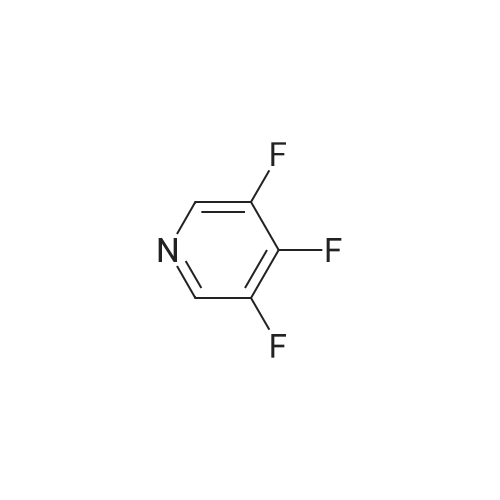
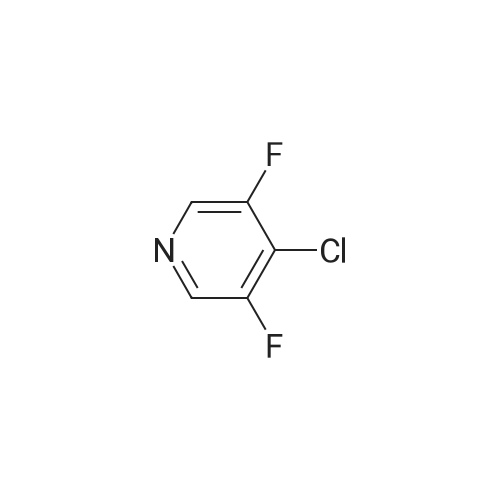
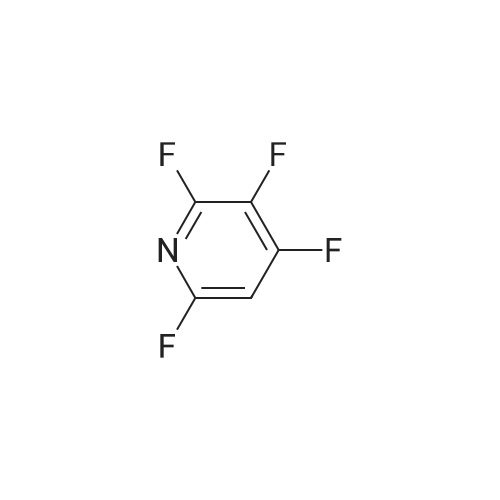
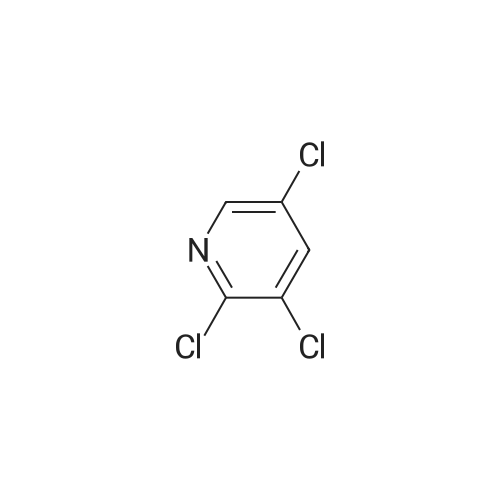
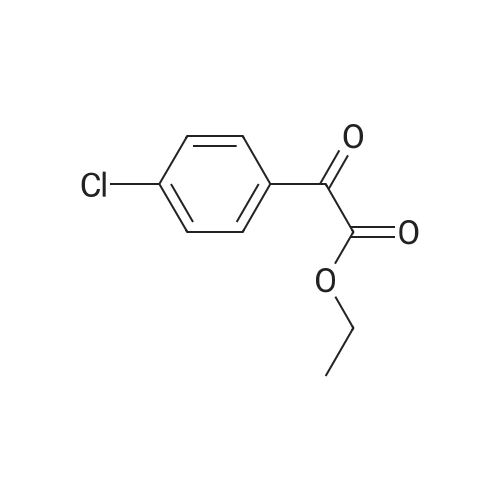
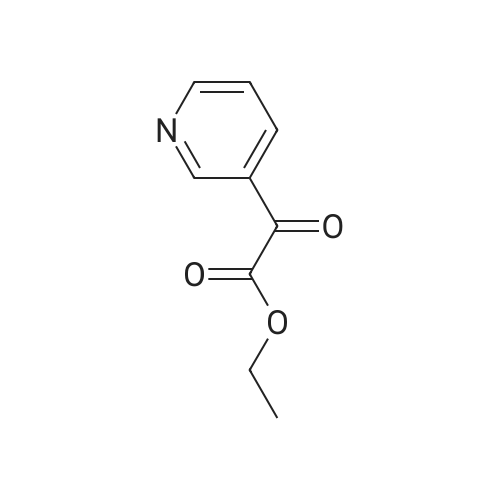
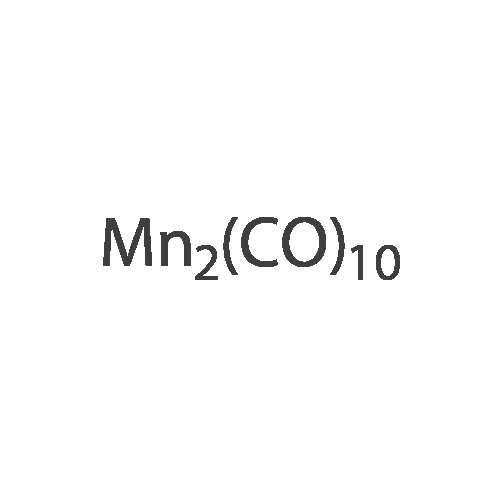There will be a HazMat fee per item when shipping a dangerous goods. The HazMat fee will be charged to your UPS/DHL/FedEx collect account or added to the invoice unless the package is shipped via Ground service. Ship by air in Excepted Quantity (each bottle), which is up to 1g/1mL for class 6.1 packing group I or II, and up to 25g/25ml for all other HazMat items.
Type
HazMat fee for 500 gram (Estimated)
Excepted Quantity
USD 0.00
Limited Quantity
USD 15-60
Inaccessible (Haz class 6.1), Domestic
USD 80+
Inaccessible (Haz class 6.1), International
USD 150+
Accessible (Haz class 3, 4, 5 or 8), Domestic
USD 100+
Accessible (Haz class 3, 4, 5 or 8), International
USD 200+
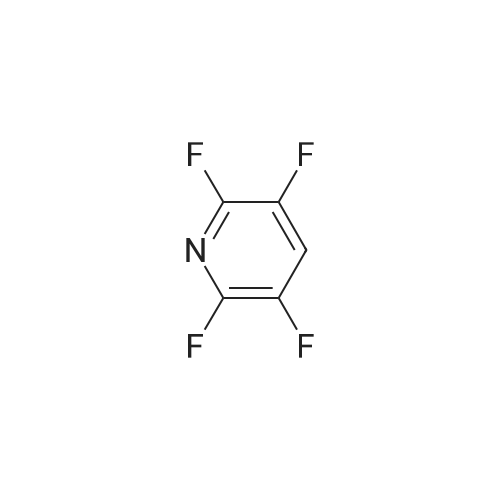
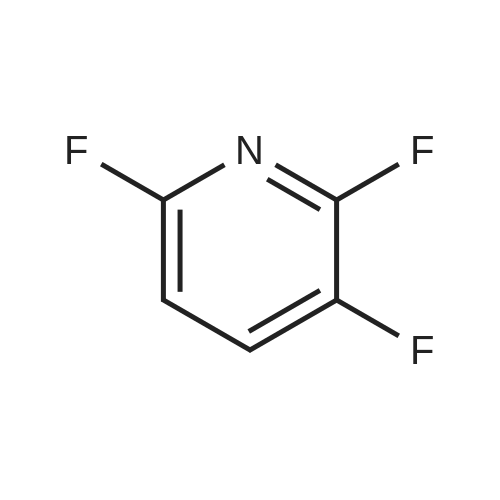
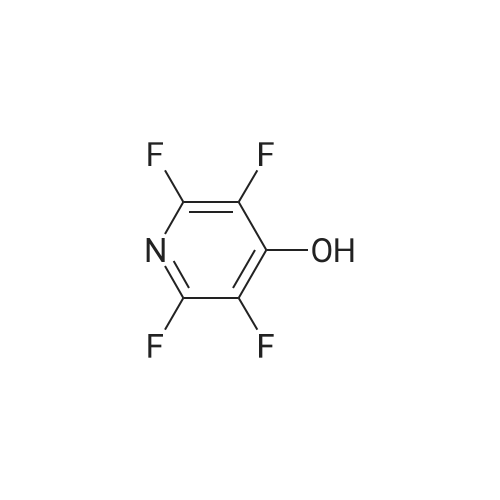
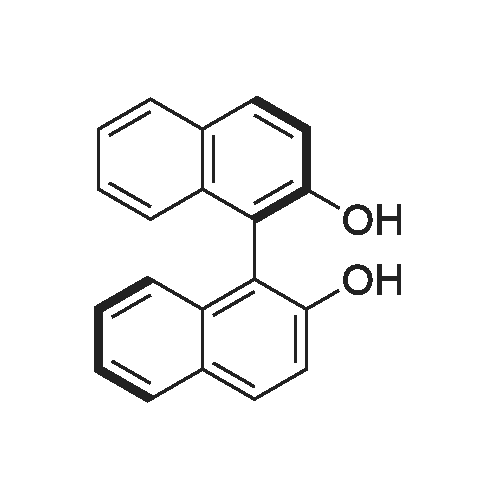
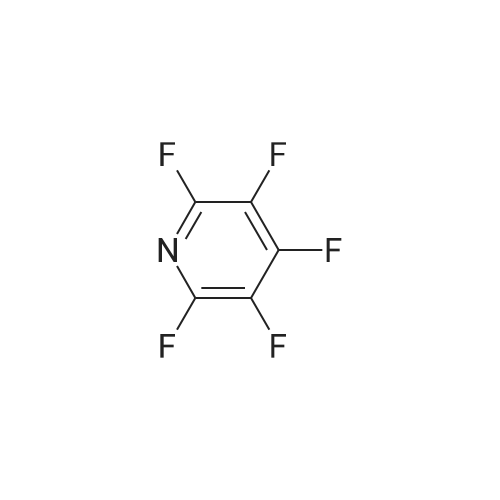
Structure of 700-16-3 * Storage: {[proInfo.prStorage]}
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
With ammonium hydroxide In tetrahydrofuran for 18 h; Reflux
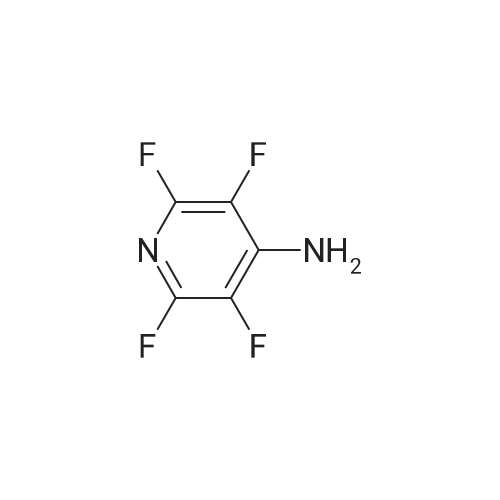
4-amino-2,3,5,6-tetrauoropyridine 1 wassynthesized from 2,3,4,5,6-Pentafluoropyridine(25 g, 148 mmol) was dissolved in THF (175 ml)in a round-bottomed ask equipped with a reuxcondenser to give a clear solution. On addition ofaqueous ammonia (0.88, 125 ml) a cloudy solutionwas produced and an exothermic reaction ensued.The mixture was then reuxed for 18 hours. Theclear solution produced was poured into water (500ml) and the whole mixture was extracted with ether(3x75ml). The extract was dried (MgSO4), evaporatedusing rotary evaporator and the residue freed fromthe last traces of solvent in vacuo, to give a palecream solid. Recrystallization of the crude materialfrom light petroleum gave long white needles of4-amino-2,3,5,6-tetrauoropyridine 1 (20 g, 0.12 mol,80percent), mp. 85–87°C; IR (KBr, cm-1): 3500-3300 (NHstr.) 1450-1190 Py-F; 1H (CDCl3): d 5.05 (2H, br s,-NH2; 19F (CDCl3): d 15.1 (2F, m, F-2 and F-6), -85.1(2F, F-3 and F-5); MS: m/z 166 ([M]+,100percent).
Reference:
[1] Journal of Fluorine Chemistry, 1996, vol. 81, # 1, p. 39 - 42
[2] Journal of the American Chemical Society, 2017, vol. 139, # 37, p. 13092 - 13101
[3] Oriental Journal of Chemistry, 2016, vol. 32, # 4, p. 1799 - 1813
[4] Journal of Organic Chemistry USSR (English Translation), 1988, vol. 24, # 12, p. 2267 - 2272[5] Zhurnal Organicheskoi Khimii, 1988, vol. 24, # 12, p. 2513 - 2518
[6] Journal of the Chemical Society, Perkin Transactions 1: Organic and Bio-Organic Chemistry (1972-1999), 1988, p. 255 - 258
[7] Journal of Fluorine Chemistry, 1982, vol. 20, p. 507 - 514
[8] Tetrahedron, 2007, vol. 63, # 30, p. 7027 - 7035
[9] Journal of Fluorine Chemistry, 2001, vol. 107, # 1, p. 13 - 22
[10] Journal of Fluorine Chemistry, 1987, vol. 36, p. 247 - 272
[11] Journal of Fluorine Chemistry, 1987, vol. 36, p. 247 - 272
[12] Journal of Fluorine Chemistry, 1987, vol. 36, p. 247 - 272
[13] Journal of Fluorine Chemistry, 1987, vol. 36, p. 247 - 272
[14] Patent: US4681873, 1987, A,
[15] Journal of the American Chemical Society, 2014, vol. 136, # 8, p. 3002 - 3005
2
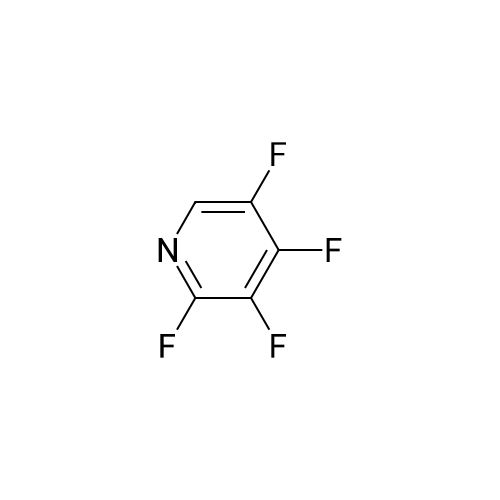
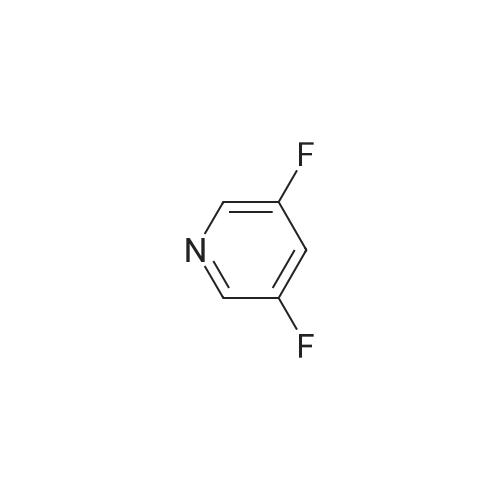
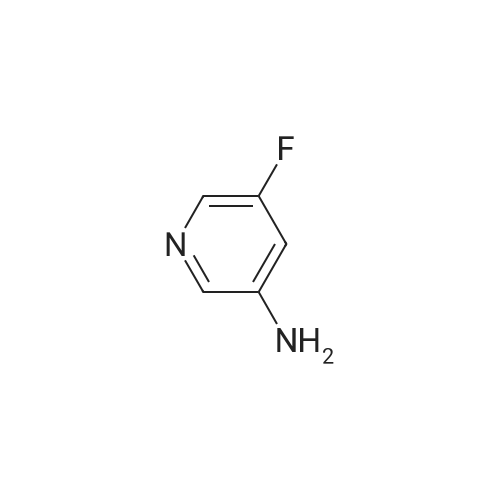
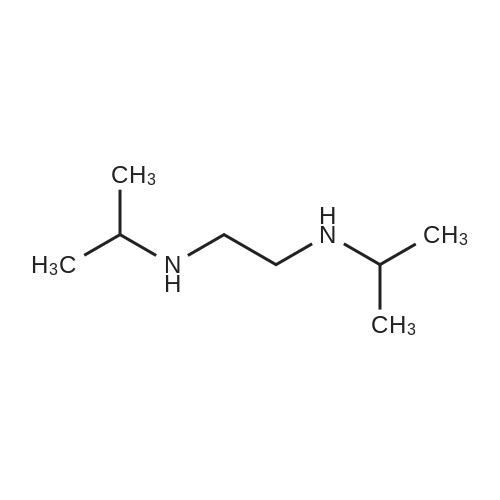
[ 700-16-3 ]
[ 35622-48-1 ]
[ 1682-20-8 ]
Reference:
[1] Journal of Fluorine Chemistry, 1995, vol. 75, # 2, p. 169 - 172
[2] Journal of Fluorine Chemistry, 1995, vol. 75, # 2, p. 169 - 172
3
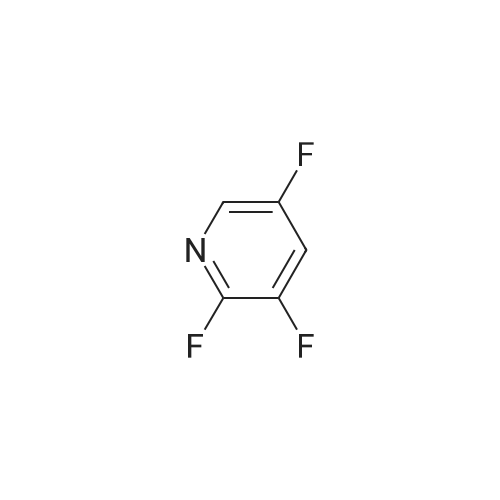
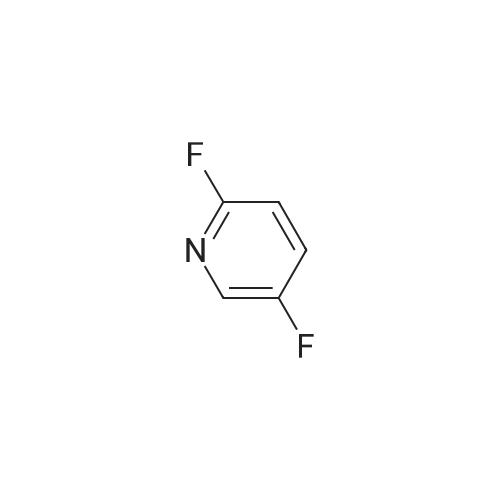
[ 700-16-3 ]
[ 4229-44-1 ]
[ 1682-20-8 ]
Reference:
[1] Journal of Fluorine Chemistry, 1987, vol. 36, p. 247 - 272
4
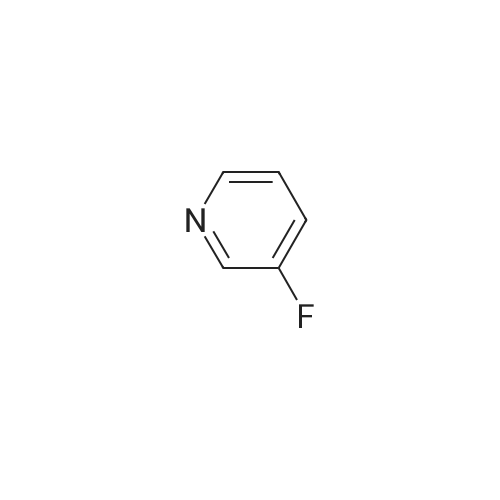
[ 700-16-3 ]
[ 7664-41-7 ]
[ 1682-20-8 ]
Reference:
[1] Journal of the Chemical Society, 1965, p. 575 - 594
[2] , Gmelin Handbook: F: PerFHalOrg.5, 4.1.1.3, page 180 - 219,
[3] C.A., 1966, vol. 65, p. 7152
[4] , Gmelin Handbook: F: PerFHalOrg.5, 4.1.1.3, page 180 - 219,
[5] Organic Letters, 2014, vol. 16, # 10, p. 2704 - 2707
[6] Chem.Abstr., 1966, vol. 65, # 7152a,
5
[ 700-16-3 ]
[ 207670-81-3 ]
[ 123394-94-5 ]
[ 1682-20-8 ]
Reference:
[1] European Journal of Inorganic Chemistry, 2001, # 8, p. 2123 - 2134
6
[ 700-16-3 ]
[ 133343-75-6 ]
[ 117847-51-5 ]
[ 1682-20-8 ]
Reference:
[1] European Journal of Inorganic Chemistry, 2001, # 8, p. 2123 - 2134
Reference:
[1] C.A., 1966, vol. 64, p. 3568
[2] C.A., 1966, vol. 65, p. 15341
[3] Chem.Abstr., 1966, vol. 65, # 15341g,
[4] , Gmelin Handbook: F: PerFHalOrg.5, 4, page 115 - 148,
[5] Chemistry and Industry (London, United Kingdom), [6] Chemistry and Industry (London, United Kingdom), 1964, p. 835 - 835
[7] , Gmelin Handbook: F: PerFHalOrg.5, 4, page 115 - 148,
[8] C.A., 1966, vol. 65, p. 7152
[9] Proceedings of the Chemical Society, London, [10] Proceedings of the Chemical Society, London, 1964, p. 83 - 83
[11] Chem.Abstr., 1966, vol. 65, # 7152a,
[12] , Gmelin Handbook: F: PerFHalOrg.5, 4, page 115 - 148,
[13] , Gmelin Handbook: F: PerFHalOrg.5, 4, page 115 - 148,
17
[ 700-16-3 ]
[ 1737-93-5 ]
[ 52026-98-9 ]
[ 52026-99-0 ]
[ 5958-25-8 ]
[ 1735-84-8 ]
Reference:
[1] Journal of Fluorine Chemistry, 1991, vol. 53, # 1, p. 33 - 42
18
[ 110-89-4 ]
[ 2176-62-7 ]
[ 700-16-3 ]
[ 1735-84-8 ]
[ 1737-93-5 ]
[ 34415-32-2 ]
Reference:
[1] Bulletin of the Academy of Sciences of the USSR, Division of Chemical Science (English Translation), 1984, vol. 33, p. 1975[2] Izvestiya Akademii Nauk SSSR, Seriya Khimicheskaya, 1984, # 9, p. 2158 - 2159
19
[ 700-16-3 ]
[ 1737-93-5 ]
[ 52026-98-9 ]
[ 52026-99-0 ]
[ 5958-25-8 ]
[ 34415-31-1 ]
[ 1735-84-8 ]
Reference:
[1] Journal of Fluorine Chemistry, 1991, vol. 53, # 1, p. 33 - 42
20
[ 2176-62-7 ]
[ 700-16-3 ]
[ 1735-84-8 ]
[ 1737-93-5 ]
Reference:
[1] C.A., 1966, vol. 64, p. 3568
[2] C.A., 1966, vol. 65, p. 15341
[3] Chem.Abstr., 1966, vol. 65, # 15341g,
[4] , Gmelin Handbook: F: PerFHalOrg.5, 4, page 115 - 148,
[5] Chemistry and Industry (London, United Kingdom), 1964, p. 835 - 835
[6] , Gmelin Handbook: F: PerFHalOrg.5, 4, page 115 - 148,
[7] C.A., 1966, vol. 65, p. 7152
[8] Proceedings of the Chemical Society, London, 1964, p. 83 - 83
[9] Chem.Abstr., 1966, vol. 65, # 7152a,
[10] , Gmelin Handbook: F: PerFHalOrg.5, 4, page 115 - 148,
[11] , Gmelin Handbook: F: PerFHalOrg.5, 4, page 115 - 148,
21
[ 110-89-4 ]
[ 2176-62-7 ]
[ 700-16-3 ]
[ 1735-84-8 ]
[ 1737-93-5 ]
[ 34415-32-2 ]
Reference:
[1] Bulletin of the Academy of Sciences of the USSR, Division of Chemical Science (English Translation), 1984, vol. 33, p. 1975[2] Izvestiya Akademii Nauk SSSR, Seriya Khimicheskaya, 1984, # 9, p. 2158 - 2159
22
[ 2176-62-7 ]
[ 700-16-3 ]
[ 1737-93-5 ]
Reference:
[1] Proceedings of the Chemical Society, London, 1964, p. 83 - 83
[2] , Gmelin Handbook: F: PerFHalOrg.5, 4, page 115 - 148,
23
[ 700-16-3 ]
[ 2176-62-7 ]
[ 52026-99-0 ]
[ 54889-43-9 ]
[ 17717-16-7 ]
[ 1737-93-5 ]
[ 51991-34-5 ]
[ 34415-32-2 ]
Reference:
[1] Journal of Fluorine Chemistry, 1991, vol. 53, # 1, p. 33 - 42
24
[ 110-86-1 ]
[ 2344-10-7 ]
[ 1513-66-2 ]
[ 700-16-3 ]
[ 371-77-7 ]
Reference:
[1] Journal of Fluorine Chemistry, 1982, vol. 21, p. 159 - 170
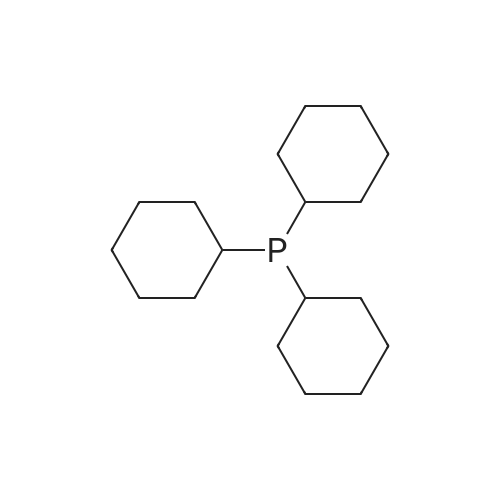
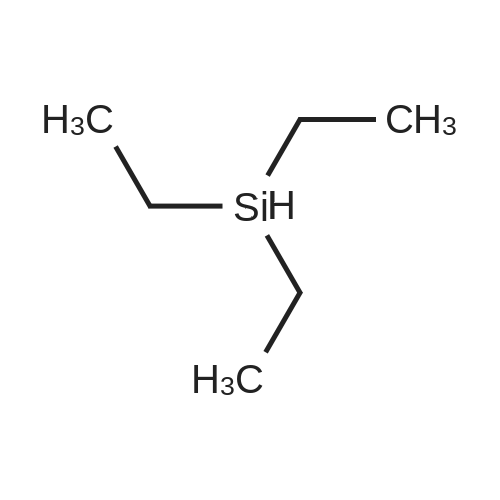
With triethylsilane; [Rh(μ-H)(1,3-bis(diisopropylphosphanyl)propane)]2 In benzene-d6 at 50℃; for 48 h; Inert atmosphere
General procedure: To a solution of fluoroarene (0.1 M) and HSiEt3 (0.1 M) in benzene-d6 in a PFA tube α,α,α-trifluorotoluene (1–2 μL) was added as internal standard. The PFA tube was closed by a Teflon plug, inserted into an NMR tube and an initial 19F{1H} NMR spectrum was recorded. Then [Rh(μ-H)(dippp)]2 (1) (0.005 M) was added and the reaction mixture was heated to 50 °C for 48 h. Hydrodefluorination of pentafluoropyridine gave 2,3,5,6-tetrafluoropyridine (11percent), 2,3,4,5-tetrafluoropyridine (11percent), 2,3,5-trifluoropyridine (8percent), 3,5-difluoropyridine (6percent) and 2-fluoropyridine (1percent) (TON = 11). Hydrodefluorination of 2,3,5,6-tetrafluoropyridine or 2,3,5,6-tetrafluoropyridine or 2,3,5,6-tetrafluoropyri-dine gave 2,3,5-trifluoropyridine (24percent), 2,3,6-trifluoropyridine (7percent), 3,5-difluoropyridine (15percent), 2,5-difluoropyridine (2percent) and 2-fluoropyridine (8percent) (TON = 18). Hydrodefluorination of hexafluoro-benzene or hexafluoroben-zene or hexa-fluorobenzene gave pentafluorobenzene (12percent) and 1,2,4,5-tetra-fluorobenzene or 1,2,4,5-tetrafluoro-benzene or 1,2,4,5-tetrafluoroben-zene (2percent) (TON = 3.1). Hydrodefluorination of pentafluorobenzene gave 1,2,4,5-tetrafluorobenzene (35percent), 1,2,3,4-tetrafluorobenzene (3percent), 1,2,4-trifluorobenzene (23percent) and 1,4-difluorobenzene (4percent) (TON = 19). Yields of organic hydrodefluorination products were determined from 19F{1H} NMR spectra by integration of product resonances versus the internal standard. Hydrodefluorination products were identified by NMR spectroscopy by comparison with literature data [23]. TON: number of hydrodefluorination steps/moles of 1.
Reference:
[1] Journal of Fluorine Chemistry, 2013, vol. 155, p. 132 - 142
Reference:
[1] Journal of the Chemical Society - Perkin Transactions 1, 1998, # 10, p. 1705 - 1713
[2] Chemistry - An Asian Journal, 2016, vol. 11, # 21, p. 3062 - 3071
Reference:
[1] Journal of Fluorine Chemistry, 1982, vol. 21, p. 159 - 170
38
[ 700-16-3 ]
[ 3512-16-1 ]
[ 2875-18-5 ]
[ 76469-41-5 ]
[ 71902-33-5 ]
Yield
Reaction Conditions
Operation in experiment
11 %Spectr.
With triethylsilane; [Rh(μ-H)(1,3-bis(diisopropylphosphanyl)propane)]2 In benzene-d6 at 50℃; for 48 h; Inert atmosphere
General procedure: To a solution of fluoroarene (0.1 M) and HSiEt3 (0.1 M) in benzene-d6 in a PFA tube α,α,α-trifluorotoluene (1–2 μL) was added as internal standard. The PFA tube was closed by a Teflon plug, inserted into an NMR tube and an initial 19F{1H} NMR spectrum was recorded. Then [Rh(μ-H)(dippp)]2 (1) (0.005 M) was added and the reaction mixture was heated to 50 °C for 48 h. Hydrodefluorination of pentafluoropyridine gave 2,3,5,6-tetrafluoropyridine (11percent), 2,3,4,5-tetrafluoropyridine (11percent), 2,3,5-trifluoropyridine (8percent), 3,5-difluoropyridine (6percent) and 2-fluoropyridine (1percent) (TON = 11). Hydrodefluorination of 2,3,5,6-tetrafluoropyridine or 2,3,5,6-tetrafluoropyridine or 2,3,5,6-tetrafluoropyri-dine gave 2,3,5-trifluoropyridine (24percent), 2,3,6-trifluoropyridine (7percent), 3,5-difluoropyridine (15percent), 2,5-difluoropyridine (2percent) and 2-fluoropyridine (8percent) (TON = 18). Hydrodefluorination of hexafluoro-benzene or hexafluoroben-zene or hexa-fluorobenzene gave pentafluorobenzene (12percent) and 1,2,4,5-tetra-fluorobenzene or 1,2,4,5-tetrafluoro-benzene or 1,2,4,5-tetrafluoroben-zene (2percent) (TON = 3.1). Hydrodefluorination of pentafluorobenzene gave 1,2,4,5-tetrafluorobenzene (35percent), 1,2,3,4-tetrafluorobenzene (3percent), 1,2,4-trifluorobenzene (23percent) and 1,4-difluorobenzene (4percent) (TON = 19). Yields of organic hydrodefluorination products were determined from 19F{1H} NMR spectra by integration of product resonances versus the internal standard. Hydrodefluorination products were identified by NMR spectroscopy by comparison with literature data [23]. TON: number of hydrodefluorination steps/moles of 1.
Reference:
[1] Journal of Fluorine Chemistry, 2013, vol. 155, p. 132 - 142
Reference:
[1] Chemistry - A European Journal, 2005, vol. 11, # 6, p. 1903 - 1910
[2] Journal of the Chemical Society - Perkin Transactions 1, 1998, # 10, p. 1705 - 1713
80 g of pentafluoropyridine (Janssen) and 111.5 g of zinc power were added to 560 ml of 20% aqueous ammonia. The resulting mixture was stirred at room temperature for 5 hours, and then gently refluxed to remove water using a Dean-Stark trap, to obtain 60.4 g (yield: 84.5%) of the title compound.
57%Chromat.
With 1,2,3,4-tetrakis(carbazol-9-yl)-5,6-dicyanobenzene; triethylamine; In 1-methyl-pyrrolidin-2-one; at 20℃; for 2h;Inert atmosphere; Irradiation;
General procedure: To a 10 mL photocatalytic reactor containing a solution of an aryl halide (0.5 mmol, 1equiv) and 5CzBN (0.01 mmol, 0.02 equiv) in dry NMP (3 mL) was added Et3N (0.8 mmol, 1.6 equiv) under nitrogen atmosphere. The solution was stirred under their radiation of 5 W blue LED for the indicated time until complete consumption of starting material as monitored by GC-MS analysis at rt. The yields were calculated from GC measurements using internal standards.
With ammonium hydroxide; In tetrahydrofuran; for 18h;Reflux;
4-amino-2,3,5,6-tetrauoropyridine 1 wassynthesized from 2,3,4,5,6-Pentafluoropyridine(25 g, 148 mmol) was dissolved in THF (175 ml)in a round-bottomed ask equipped with a reuxcondenser to give a clear solution. On addition ofaqueous ammonia (0.88, 125 ml) a cloudy solutionwas produced and an exothermic reaction ensued.The mixture was then reuxed for 18 hours. Theclear solution produced was poured into water (500ml) and the whole mixture was extracted with ether(3x75ml). The extract was dried (MgSO4), evaporatedusing rotary evaporator and the residue freed fromthe last traces of solvent in vacuo, to give a palecream solid. Recrystallization of the crude materialfrom light petroleum gave long white needles of4-amino-2,3,5,6-tetrauoropyridine 1 (20 g, 0.12 mol,80%), mp. 85-87C; IR (KBr, cm-1): 3500-3300 (NHstr.) 1450-1190 Py-F; 1H (CDCl3): d 5.05 (2H, br s,-NH2; 19F (CDCl3): d 15.1 (2F, m, F-2 and F-6), -85.1(2F, F-3 and F-5); MS: m/z 166 ([M]+,100%).
With ammonia;
A. 4-Amino-2,3,5,6-tetrafluoropyridine A mixture of 20.0 g (118 mmol) of pentafluoropyridine (Aldrich Chemical Company, Milwaukee, Wis., USA) and 40 ml of concentrated aqueous ammonia was heated on a steam bath for two hours in a sealed stainles steel reaction vessel. The vessel and contents were cooled to room temperature and the resulting suspension suspension was partitioned between water and diethyl ether. The water layer was extracted with ether and the ether solutions were combined and dried over anhydrous magnesium sulfate. Evaporation of the ether yielded 16.3 g (83.0%) of the title compound as a white solid of sufficient purity for use in the next reaction step. The material exhibited an Rf value of 0.4 when chromatographed on a thin-layer silica gel plate using chloroform as the eluant. A minor impurity (Rf =0.2) was also observed. Recrystallization of the reaction product from cyclohexane provided pure material, mp 80-82 C. (sublimation).
With tetrabutylammomium bromide In N,N-dimethyl acetamide at 100℃; for 20h; Inert atmosphere;
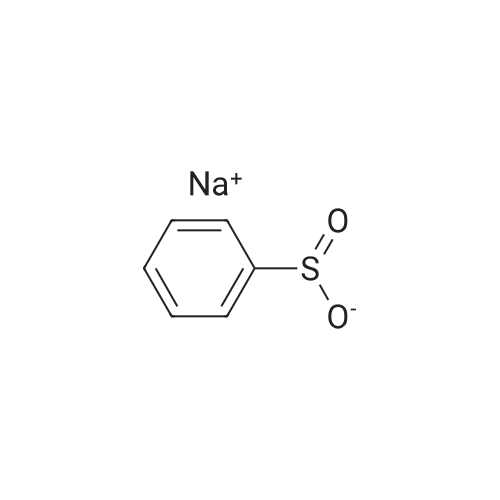
1
Under the protection of nitrogen, dissolve sodium benzenesulfinate (1.94g, 11.83mmol), pentafluoropyridine (2.00g, 11.83mmol) and tetrabutylammonium bromide (1.14g, 3.54mmol) in 10mL N, N- Dimethylacetamide was stirred at 100°C (600 rpm) for 20 hours.After the reaction solution was cooled to room temperature, 30 mL of deionized water was slowly added, filtered with suction, and washed with water three times to obtain the product.The product was further separated and purified by a silica gel chromatographic column and dried in vacuum to obtain probe A (2.70 g, 78.37%).
In N,N-dimethyl-formamide Reflux; regioselective reaction;
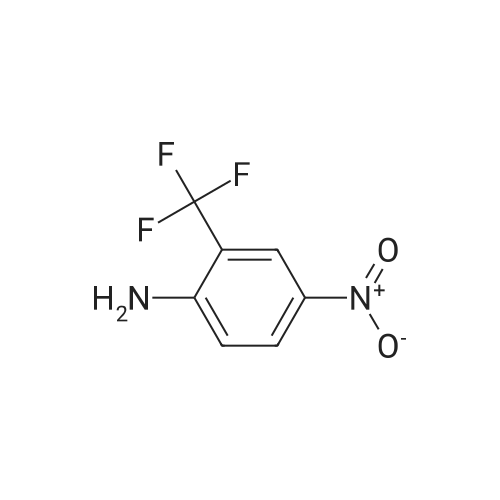
EXAMPLE 2 PREPARATION OF N-(2,3,5,6-TETRAFLUORO-4-PYRIDYL)-N-(4-NITRO-2-TRIFLUOROMETHYLPHENYL)AMINE Sodium hydride (2.0 grams of 50% oil dispersion) was washed with pentane, filtered, and cooled to 0 C. A solution of 4-nitro-2-trifluoromethylaniline (6.1 grams; 0.030 mole) in 25 ml. of DMF was added over a five-minute period. The reaction mixture was allowed to stir for one hour, the temperature rising as high as 10 C. The reaction mixture was then cooled back down to 0 C. and pentafluoropyridine (5.0 grams; 0.030 mole) in 25 ml. of DMF was added. The reaction mixture was allowed to stir without further cooling for about sixteen hours, then poured over 1.8 liter of crushed ice and the volume adjusted to 1.8 liter with water. A precipitate formed and was allowed to remain in the reaction mixture for about twenty-four hours. The precipitate was then separated by filtration. It was recrystallized from ethanol, yielding 2.6 grams of a first crop (m.p. 104-5 C.; U.S. Pat. No. 3,926,611 reports 107.7-108) and 3.0 grams of a second crop. Elemental analysis of the first crop showed Calc.: C, 40.58; H, 1.14; N, 11.83. Found: C, 40.34; H, 1.36; N, 11.83.
4-(1-(pyridine-4-yl)vinyloxy)-2,3,5,6-tetrafluoropyridine[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
55%
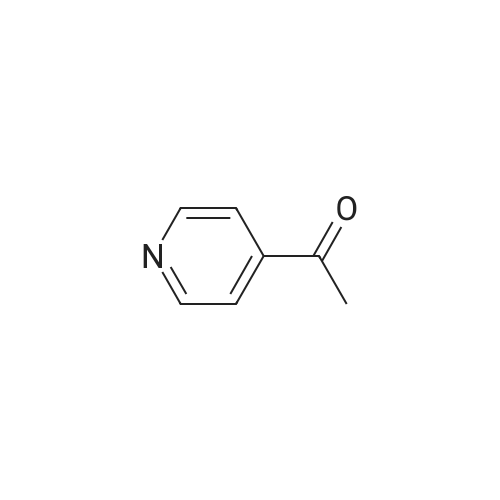
Stage #1: methyl (4-pyridyl) ketone With sodium hydride In tetrahydrofuran at 20℃; for 1.5h; Inert atmosphere;
Stage #2: Pentafluoropyridine In tetrahydrofuran at 80℃; for 12h; Inert atmosphere;
4.1 RRN General procedure for reactions between pentafluoropyridine and aromatic and heteroaromatic ketones
General procedure: Under an atmosphere of dry nitrogen, sodium hydride (1.5mmol) was added to a solution of ketone (1mmol) in dry THF (10ml) and the mixture was stirred at room temperature for 90min. Then pentafluoropyridine 1 (1mmol) was added and resulting solution was refluxed at 80°C for 12h. The reaction mixture was cooled to room temperature and the solvent was evaporated. The reaction mixture was poured onto 0.2M hydrochloric acid (30ml), and then extracted with chloroform, dried (MgSO4) and evaporated to yield the crude product, which was then purified by column chromatography on silica gel.
With triethylsilane; [Rh(mu-H)(1,3-bis(diisopropylphosphanyl)propane)]2; In benzene-d6; at 50℃; for 48h;Inert atmosphere;
General procedure: To a solution of fluoroarene (0.1 M) and HSiEt3 (0.1 M) in benzene-d6 in a PFA tube alpha,alpha,alpha-trifluorotoluene (1?2 muL) was added as internal standard. The PFA tube was closed by a Teflon plug, inserted into an NMR tube and an initial 19F{1H} NMR spectrum was recorded. Then [Rh(mu-H)(dippp)]2 (1) (0.005 M) was added and the reaction mixture was heated to 50 °C for 48 h. Hydrodefluorination of pentafluoropyridine gave 2,3,5,6-tetrafluoropyridine (11percent), 2,3,4,5-tetrafluoropyridine (11percent), 2,3,5-trifluoropyridine (8percent), 3,5-difluoropyridine (6percent) and 2-fluoropyridine (1percent) (TON = 11). Hydrodefluorination of 2,3,5,6-tetrafluoropyridine or 2,3,5,6-tetrafluoropyridine or 2,3,5,6-tetrafluoropyri-dine gave 2,3,5-trifluoropyridine (24percent), 2,3,6-trifluoropyridine (7percent), 3,5-difluoropyridine (15percent), 2,5-difluoropyridine (2percent) and 2-fluoropyridine (8percent) (TON = 18). Hydrodefluorination of hexafluoro-benzene or hexafluoroben-zene or hexa-fluorobenzene gave pentafluorobenzene (12percent) and 1,2,4,5-tetra-fluorobenzene or 1,2,4,5-tetrafluoro-benzene or 1,2,4,5-tetrafluoroben-zene (2percent) (TON = 3.1). Hydrodefluorination of pentafluorobenzene gave 1,2,4,5-tetrafluorobenzene (35percent), 1,2,3,4-tetrafluorobenzene (3percent), 1,2,4-trifluorobenzene (23percent) and 1,4-difluorobenzene (4percent) (TON = 19). Yields of organic hydrodefluorination products were determined from 19F{1H} NMR spectra by integration of product resonances versus the internal standard. Hydrodefluorination products were identified by NMR spectroscopy by comparison with literature data [23]. TON: number of hydrodefluorination steps/moles of 1.
N-(2,3,5,6-tetrafluoropyridine)dehydrobutyrine methyl ester[ No CAS ]
[ 1612240-79-5 ]
[ 1612240-80-8 ]
Yield
Reaction Conditions
Operation in experiment
2%; 50%; 9%
With potassium carbonate; In acetonitrile; at 20℃; for 18h;
General procedure: Reaction of threonine derivative with pentafluoropyridine (PFP) in the presence of potassium carbonate PFP (2 equiv) was added to a stirred suspension of amino acid starting material (200 mg, 1 equiv), potassium carbonate (3 equiv) and acetonitrile (8 mL) and left to stir at room temperature for 18 h and purified as indicated below.
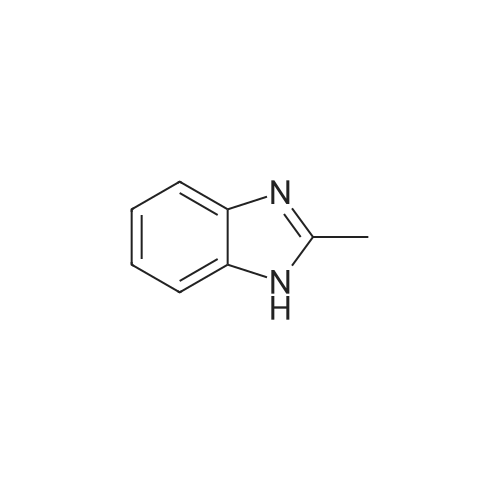
1-(2,3,5,6-tetrafluoropyridyl)-2-methylbenzimidazole[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
84.9%
In tetrahydrofuran at 20℃; for 408h;
4.6. Preparation of 1-(2,3,5,6-tetrafluoropyridyl)-2-methylbenzimidazole (4)
2-Methylbenzimidazole (0.504 g, 3.81 mmol) in tetrahydrofuran(50 cm3) was added to pentafluoropyridine (0.598 g,3.54 mmol) and solution left at ambient temperature for 17 days. The solvent was removed by rotary evpaoration and the product extracted into dichloromethane (2 50 cm3) and filtered. The solvent was removed by rotary evaporation to afford the product 4 as a white solid. Yield: 0.842 g (84.9%). Anal. Calcd. for C13H7F4N3:C, 55.5; H, 2.5; N, 14.9. Found: C, 55.5; H, 2.6; N 15.0%. MS:C13H8F4N3 requires 282.0654; found [M + H+] 282.0659. 1H NMR:d = 7.85 (1H, d, J = 7.5 Hz, C6H4), 7.41 (1H, ddd, J = 7.5 Hz, J = 7.5 Hz,J = 1.0 Hz, C6H4), 7.36 (1H, ddm, J = 7.5 Hz, J = 7.5 Hz, C6H4), 7.12(1H, d, J = 7.5 Hz, C6H4), 2.60 (s, CH3). 13C{1H} NMR: d = 151.6 (s,N2C), 144.2 (dm, 1JCF = 251 Hz, C5F4N), 142.9 (s, C6H4), 138.1 (dm,1JCF = 251 Hz, C5F4N), 134.5 (s, C6H4), 127.3 (t, JCF = 13 Hz, C5F4N),124.1 (s, C6H4), 124.0 (s, C6H4), 120.0 (s, C6H4), 109.5 (s, C6H4), 13.9(s, CH3). 19F NMR: d = 85.72 (2F), 143.68 (2F) (A and Bcomponents of an AA’BB’ spin pattern).
84.9%
In tetrahydrofuran at 20℃; for 408h;
4.6. Preparation of 1-(2,3,5,6-tetrafluoropyridyl)-2-methylbenzimidazole (4)
2-Methylbenzimidazole (0.504 g, 3.81 mmol) in tetrahydrofuran (50 cm3) was added to pentafluoropyridine (0.598 g, 3.54 mmol) and solution left at ambient temperature for 17 days. The solvent was removed by rotary evpaoration and the product extracted into dichloromethane (2 x 50 cm3) and filtered. The solvent was removed by rotary evaporation to afford the product 4 as a white solid. Yield: 0.842 g (84.9%).
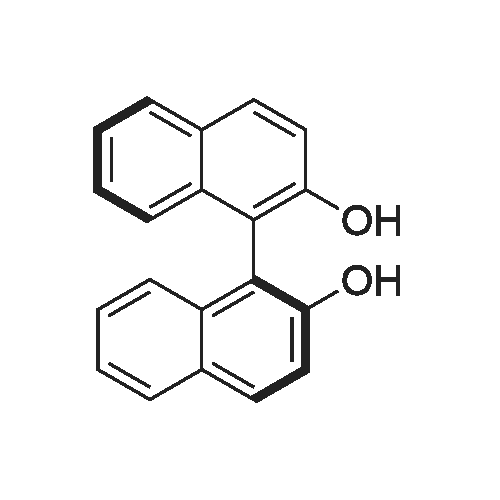
(R)-2,2'-bis(tetrafluoropyridin-4-yloxy)-1,1'-binaphthyl[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
95%
With potassium carbonate In N,N-dimethyl-formamide at 20℃;
(R,S)-2,2'-bis(tetrafluoropyridin-4-yloxy)-1,1'-binaphthyl 2 (typicalprocedure).
General procedure: PFP (2.48 g, 14.7 mmol) was added dropwise to a stirred mixture of racemic 1 (2 g, 7 mmol) and powdered K2CO3 (1.93 g, 14 mmol) in 15 ml of DMF. The progress of the reaction was followed by TLC (eluent: benzene-acetone, 10:1) and when traces of 1 and intermediate 3 were disappeared (in 15-20 min), the resulting mixture was poured into water (100 ml). The precipitate (white crystals) was filtered off, washed with water and dried to obtain racemic 2.
(S)-2,2'-bis(tetrafluoropyridin-4-yloxy)-1,1'-binaphthyl[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
95%
With potassium carbonate In N,N-dimethyl-formamide at 20℃;
(R,S)-2,2'-bis(tetrafluoropyridin-4-yloxy)-1,1'-binaphthyl 2 (typicalprocedure).
General procedure: PFP (2.48 g, 14.7 mmol) was added dropwise to a stirred mixture of racemic 1 (2 g, 7 mmol) and powdered K2CO3 (1.93 g, 14 mmol) in 15 ml of DMF. The progress of the reaction was followed by TLC (eluent: benzene-acetone, 10:1) and when traces of 1 and intermediate 3 were disappeared (in 15-20 min), the resulting mixture was poured into water (100 ml). The precipitate (white crystals) was filtered off, washed with water and dried to obtain racemic 2.
[R]-2′-[(2,3,5,6-tetrafluoropyridin-4-yl)oxy]-[1,1′-binaphthalene]-2-ol[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
45%
With potassium carbonate In N,N-dimethyl-formamide at 20℃; for 1h;
(R,S)-2,2'-bis(tetrafluoropyridin-4-yloxy)-1,1'-binaphthyl 2 (typicalprocedure).
General procedure: PFP (2.48 g, 14.7 mmol) was added dropwise to a stirred mixture of racemic 1 (2 g, 7 mmol) and powdered K2CO3 (1.93 g, 14 mmol) in 15 ml of DMF. The progress of the reaction was followed by TLC (eluent: benzene-acetone, 10:1) and when traces of 1 and intermediate 3 were disappeared (in 15-20 min), the resulting mixture was poured into water (100 ml). The precipitate (white crystals) was filtered off, washed with water and dried to obtain racemic 2.
With potassium carbonate In water at 20℃; for 4h; Green chemistry;
General procedure for preparation of compound 3a-i
General procedure: Potassium carbonate (0.2 mmol) was added to the mixture of nucleophile (0.1 mmol) in water (3 mL). Then pentafluoropyridine or tetrafluoropyrimidine (0.1 mmol for 3a and 0.2 mmol for other compounds) was added and the resulting solution was stirred at room temperature for a few hours. After completion of the reaction as indicated by TLC, CHCl3 (20 mL) and brine (20 mL) were added then the organic layer collected. The CHCl3 was evaporated and column chromatography on silica gel using EtOAc-hexane (1:5)as eluent (for compound 3f and 3h) or recrystallization from EtOAc (for all compounds except 3f and 3h) gave the product.
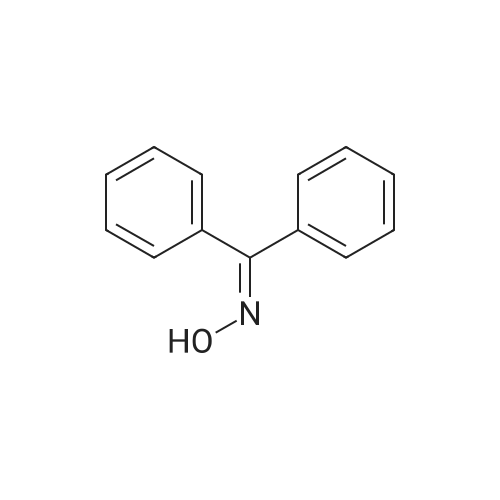
diphenylmethanone O-(2,3,5,6-tetrafluoro-4-pyridinyl)oxime[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
95%
Stage #1: Benzophenone oxime With sodium hydrogencarbonate In acetonitrile at 20℃; for 0.25h;
Stage #2: Pentafluoropyridine In acetonitrile at 87℃; for 48h; Reflux;
Diphenylmethanone O-(2,3,5,6-tetrafluoro-4-pyridinyl)-oxime
General procedure: Sodium hydrogen carbonate (1 mmol) was added to themixture of oxime (2, 1 mmol) in 10 cm3 acetonitrile. Themixture was stirred at room temperature for 15 min.Afterwards pentafluoropyridine (1, 1 mmol) was added andthe resulting solution was refluxed at 87 C for 48 h. Aftercompletion of the reaction, as indicated by TLC (ethyl acetate/n-hexane 2:5), the reaction mixture was cooled toroom temperature and the solvent was evaporated. Thereaction mixture was poured onto 15 cm3 0.5 M hydrochloricacid and 10 cm3 water was added; thenextracted with dichloromethane, dried (MgSO4), andevaporated to yield the crude product, which was thenpurified by recrystallization from diethyl ether/petroleumether. The products were characterized by conventionalspectroscopic methods. Pale yellow solid; yield: 95 %; m.p.: 157-159 C; IR(KBr): m = 3054, 1655, 1599, 1534, 1438, 1322, 750, 715,690 cm-1; 1H NMR (400 MHz, CDCl3): d = 7.18 (t,J = 7.6 Hz, 1H, Ar-H), 7.39 (t, J = 8.0 Hz, 2H, Ar-H),7.50 (t, J = 8.0 Hz, 2H, Ar-H), 7.56 (t, J = 5.2 Hz, 1H,Ar-H), 7.66 (d, J = 7.6 Hz, 2H, Ar-H), 7.89 (d,J = 7.2 Hz, 2H, Ar-H) ppm; 19F NMR (376 MHz,CDCl3): d = -154.91 (d, J = 3.7 Hz, F-3), -89.99 (dd,J = 18.8, 15.0 Hz, F-2) ppm; 13C NMR (100 MHz,CDCl3): d = 120.2, 124.6, 127.0, 128.8, 129.1, 131.8,135.0, 137.9, 165.7 ppm; MS (EI, 70 eV): m/z (%) = 346(M?, 1), 320 (60), 294 (3), 258 (51), 236 (7), 202 (73), 171(14), 140 (100), 105 (52), 83 (27), 43 (52).
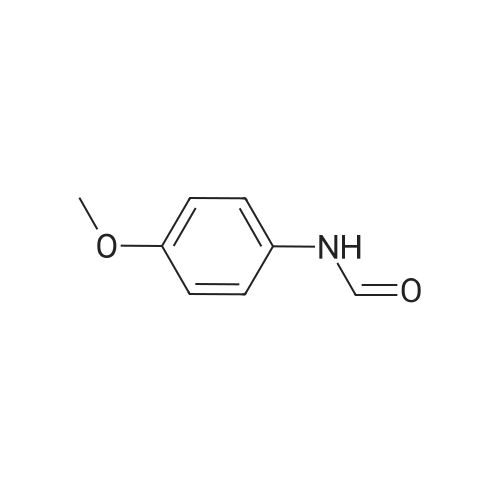
2,3,5,6-tetrafluoropyridin-4-yl-N-(4-methoxyphenyl)-formimidate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
30%
Stage #1: 4-methoxyformanilide With potassium carbonate In tetrahydrofuran at 20℃; for 0.5h;
Stage #2: Pentafluoropyridine In tetrahydrofuran at 70℃; for 20h; chemoselective reaction;
4.3. General procedure for reactions between pentafluoropyridine andN-aryl formamides
General procedure: Potassium carbonate (1.5 mmol) was added to a solution offormamide 3 (1 mmol) in dry THF (5 ml) and the mixture wasstirred at room temperature for 30 min. Then, pentafluoropyridine4 (1 mmol) was added and the resulting solution was refluxed at70 8C for 20 h. The reaction mixture was poured on 10 ml waterand extracted with ethylacetate (3 8 ml), then dried with MgSO4and solvent evaporated. The product was obtained after purificationwith column chromatography.
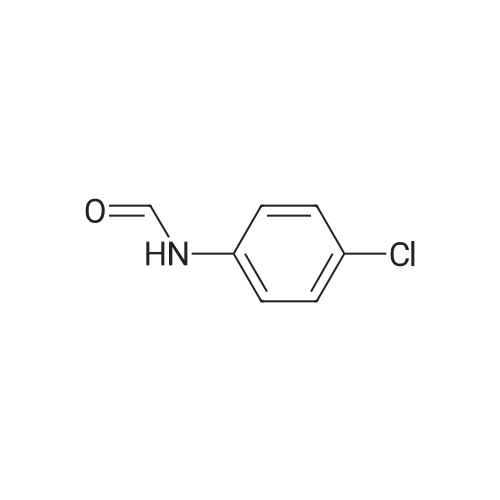
2,3,5,6-tetrafluoropyridin-4-yl-N-(4-chlorophenyl)formimidate[ No CAS ]
N-(4-chlorophenyl)-2,3,5,6-tetrafluoropyridin-4-amine[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
1: 15%
2: 40%
Stage #1: N-(4-chlorophenyl)formamide With potassium carbonate In tetrahydrofuran at 20℃; for 0.5h;
Stage #2: Pentafluoropyridine In tetrahydrofuran at 70℃; for 20h; chemoselective reaction;
4.3. General procedure for reactions between pentafluoropyridine andN-aryl formamides
General procedure: Potassium carbonate (1.5 mmol) was added to a solution offormamide 3 (1 mmol) in dry THF (5 ml) and the mixture wasstirred at room temperature for 30 min. Then, pentafluoropyridine4 (1 mmol) was added and the resulting solution was refluxed at70 8C for 20 h. The reaction mixture was poured on 10 ml waterand extracted with ethylacetate (3 8 ml), then dried with MgSO4and solvent evaporated. The product was obtained after purificationwith column chromatography.
4-bromo-N′-((perfluoropyridin-4-yl)oxy)benzimidamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
77%
Stage #1: p-bromobenzamidoxime With potassium carbonate In acetonitrile at 20℃; for 0.5h;
Stage #2: Pentafluoropyridine In acetonitrile for 6h; Reflux;
General procedure for reactions between pentafluoropyridine and amidoximes 4a-g
General procedure: Potassium carbonate (3 mmol) was added to a solution of amidoxime 2 (1 mmol) in acetonitrile (5 mL), and the mixture was stirred at room temperature for 30 min. Then, pentafluoropyridine 3 (1 mmol) was added, and the resulting solution was refluxed for 6 h. The reaction mixture was poured into water (10 mL) and extracted with CHCl3(3 × 10 mL) and dried over MgSO4 and the solvent evaporated. The product was obtained after purification with preparative thin-layer chromatography (EtOH/EtOAc).
4-bromo-N-(perfluoropyridin-4-yl)-N-((perfluoropyridin-4-yl)oxy)benzimidamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
Stage #1: p-bromobenzamidoxime With potassium carbonate In acetonitrile at 20℃; for 0.5h;
Stage #2: Pentafluoropyridine In acetonitrile for 7h; Reflux;
General procedure for synthesis of N-(perfluoropyridin-4-yl)-N-((perfluoropyridin-4-yl)oxy)imidamide systems 4j, 4k
General procedure: Potassium carbonate (3 mmol) was added to a solution of amidoxime 2 (1 mmol) in acetonitrile (5 mL), and the mixture was stirred at room temperature for 30 min. Then, pentafluoropyridine 3 (2 mmol) was added, and the resulting solution was refluxed for 7 h. The reaction mixture was poured into 10 mL of water and extracted with CHCl3 (3 × 10 mL) and dried over MgSO4 and the solvent evaporated. The product was obtained after purification with preparative thin-layer chromatography (EtOH/EtOAc).
N′-((perfluoropyridin-4-yl)oxy)nicotinimidamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
62%
General procedure: Potassium carbonate (3 mmol) was added to a solution of amidoxime 2 (1 mmol) in acetonitrile (5 mL), and the mixture was stirred at room temperature for 30 min. Then, pentafluoropyridine 3 (1 mmol) was added, and the resulting solution was refluxed for 6 h. The reaction mixture was poured into water (10 mL) and extracted with CHCl3(3 × 10 mL) and dried over MgSO4 and the solvent evaporated. The product was obtained after purification with preparative thin-layer chromatography (EtOH/EtOAc).
5,7,8-trifluoro-3-(pyridin-2-yl)-4H-pyrido[3,4-e][1,2,4]oxadiazine[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
General procedure: Potassium carbonate (3 mmol) was added to a solution of amidoxime 2 (1 mmol) in acetonitrile (20 mL), and the mixture was stirred at room temperature for 30 min. Then, pentafluoropyridine 3 (1 mmol) was added, and the resulting solution was refluxed for 24 h. The reaction mixture was poured into 10 mL of water and extracted with CHCl3 (3 × 10 mL) and dried over MgSO4 and the solvent evaporated. The product was obtained after purification with preparative thin-layer chromatography (EtOH/EtOAc).
2',3',5',6'-tetrafluoro-4H-1,4'-bipyridin-4-one[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
90%
General procedure: K2CO3 (207 mg, 1.5 mmol) was added to a solution of pyridinol 3(1 mmol) in dry THF (5 ml), and the mixture was stirred at room temperature for 30 min. Then pentafluoropyridine (1)(169 mg, 1 mmol) or pentachloropyridine (2) (253 mg,1 mmol) was added and the resulting solution was stirredfor 6 h at room temperature. After completion of the reaction (indicated by TLC), the solvent was evaporated,and products were obtained after purification using column chromatography with EtOAc-CHCl3, 4:1. 2',3',5',6'-Tetrafluoro-4H-1,4'-bipyridin-4-one (4a).Pyridin-4-ol (3a) (95 mg, 1 mmol) was used. Yield 219 mg(90%), white crystals, mp 179C. IR spectrum, nu, cm-1: 3097 (C-H), 1632 (C=O), 1586 (C=N), 1573 (C=C). 1H NMR spectrum (300 MHz, CDCl3), delta, ppm (J, Hz): 6.65(2H, d, 3JHH = 6.6, H pyridone); 7.39-7.58 (2H, m,H pyridone). 13C NMR spectrum (75 MHz, CDCl3), delta, ppm(J, Hz): 120.0; 138.5; 140.1 (dm, 1JCF = 251); 142.5; 145.3(dm, 1JCF = 252); 178.2. 19F NMR spectrum (282 MHz,CDCl3), delta, ppm: -148.48÷-148.37 (2F, m, m-F); -84.49÷-84.32 (2F, m, o-F). Found, %: C 49.05; H 1.52; N 11.30.C10H4F4N2O. Calculated, %: C 49.19; H 1.65; N 11.47.
(3) Using pentafluoropyridine as a raw material and DMF as a solvent, the active potassium fluoride powder is a fluorinating agent under the action of an excessive amount of nano-zinc-containing layered manganese oxide catalyst under a strong alkaline condition with a pH of 14. ,20 reaction for 20 h, filtered, and the filter cake was added to a solution of calcium chloride,2,3,5-Trichloropyridine is obtained based on a nanozinc-containing layered manganese oxide catalyst.
With hydrogen; potassium carbonate; at 200℃; under 15001.5 Torr; for 2h;Autoclave;
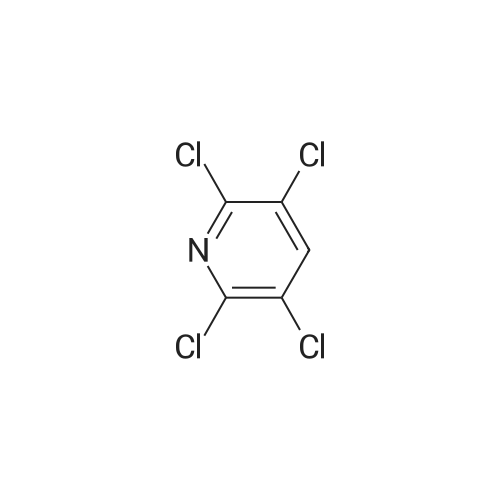
1. In a 250 ml high pressure reactor, 40 g of pentachloropyridine, 0.5 g of Raney nickel (catalyst mass ratio of 80:1), 44 g of potassium carbonate (pentachloropyridine and acid binding agent molar ratio of 1:2) were placed, and the high pressure reactor was closed. 2. Replace the air in the kettle with hydrogen and refill the hydrogen to a pressure of 2 MPa in the kettle. The temperature was raised to 200 C and kept for 0.5 hours. 3. Online monitoring of the content of tetrachloropyridine in the kettle is 50.1% (the tetrachloro method used in this paper is the liquid phase external standard), and it is cooled to room temperature, and the acid gas in the kettle is discharged under pressure. 4. Refill the hydrogen to the pressure in the kettle to 2 MPa. The temperature was raised to 200 C and kept for 0.5 hours. 5. Online monitoring of the content of tetrachloropyridine in the kettle was 75.5%, and it was cooled to room temperature, and the acid gas in the kettle was discharged under pressure. 6. Refill the hydrogen to the pressure in the kettle to 2 MPa. The temperature was raised to 200 C and kept for 0.5 hours. 7. On-line monitoring of the content of tetrachloropyridine in the kettle was 87.2%, and it was cooled to room temperature, and the acid gas in the kettle was discharged under pressure. 8. Refill the hydrogen to a pressure of 2 MPa in the kettle. Warm to 200 C and keep warm for 0.5 hours. 9. On-line monitoring of the content of tetrachloropyridine in the kettle was 92.5%, and it was lowered to room temperature, and the acid gas in the kettle was discharged under pressure to terminate the reaction. 10. Add petroleum ether to the autoclave, stir for 20 minutes, filter by suction, concentrate the filtrate at 30 C under reduced pressure to a certain extent, add water, stir at 35 C for 10 h, then naturally cool to room temperature, suction filtration, filter cake scraping Drying at 60 C gives 2,3,5,6-tetrachloropyridine (purity 99.2%).
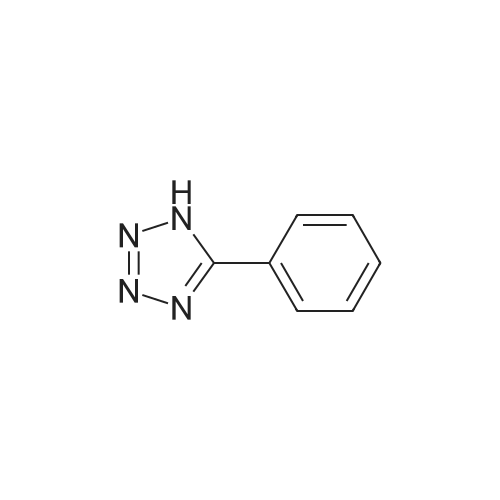
2,3,5,6-tetrafluoro-4-(5-phenyl-2H-tetrazol-2-yl)pyridine[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
85%
With potassium carbonate In acetonitrile at 20℃; for 2h; regioselective reaction;
4.1 General procedure for reaction of pentafluoropyridine and tetrazoles
General procedure: A mixture of tetrazole derivative (1 mmol) and pentafluoropyridine (1 mmol for 1a-e, 1 h and 1i; 2 mmol for 1f and 1 g) at the presence of potassium carbonate (1.5 mmol for 1a-e, 1 h and 1i; 3 mmol for 1f and 1 g) in 5 ml acetonitrile was stirred at room temperature for 2 h (4 h for 1f and 1 g). The reaction mixture was poured into 10 ml of water and obtained precipitate was filtered. In case precipitate was not formed, the mixture was extracted with ethyl acetate (3 5 ml), organic phase dried with MgSO4 and the solvent evaporated. The product was obtained after recrystallization with ethanol or purification with column chromatography (n-hexane/EtOAc).
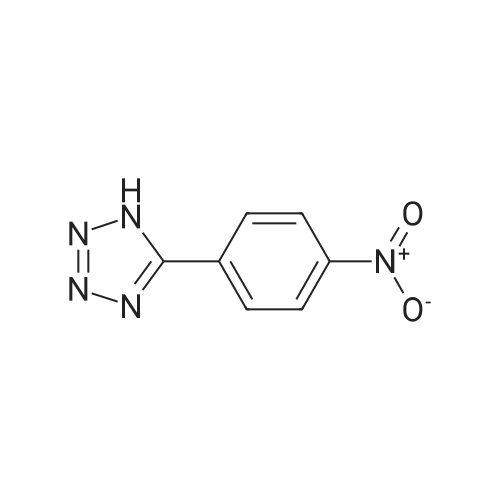
2,3,5,6-tetrafluoro-4-(5-(4-nitrophenyl)-2H-tetrazol-2-yl)pyridine[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
78%
With potassium carbonate; In acetonitrile; at 20℃; for 2h;
General procedure: A mixture of tetrazole derivative (1 mmol) and pentafluoropyridine (1 mmol for 1a-e, 1 h and 1i; 2 mmol for 1f and 1 g) at the presence of potassium carbonate (1.5 mmol for 1a-e, 1 h and 1i; 3 mmol for 1f and 1 g) in 5 ml acetonitrile was stirred at room temperature for 2 h (4 h for 1f and 1 g). The reaction mixture was poured into 10 ml of water and obtained precipitate was filtered. In case precipitate was not formed, the mixture was extracted with ethyl acetate (3 5 ml), organic phase dried with MgSO4 and the solvent evaporated. The product was obtained after recrystallization with ethanol or purification with column chromatography (n-hexane/EtOAc).
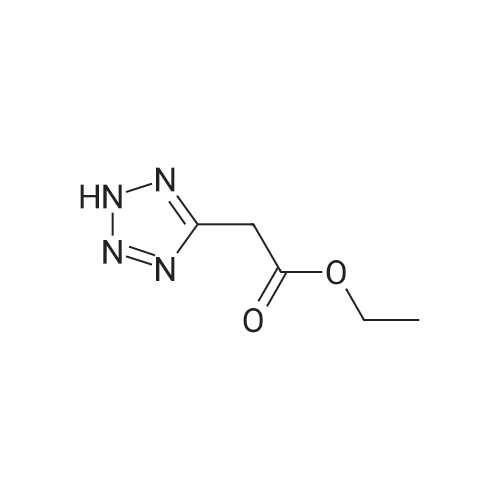
ethyl 2-(2-(2,3,5,6-tetrafluoropyridin-4-yl)-2H-tetrazol-5-yl)acetate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
83%
With potassium carbonate; In acetonitrile; at 20℃; for 2h;
General procedure: A mixture of tetrazole derivative (1 mmol) and pentafluoropyridine (1 mmol for 1a-e, 1 h and 1i; 2 mmol for 1f and 1 g) at the presence of potassium carbonate (1.5 mmol for 1a-e, 1 h and 1i; 3 mmol for 1f and 1 g) in 5 ml acetonitrile was stirred at room temperature for 2 h (4 h for 1f and 1 g). The reaction mixture was poured into 10 ml of water and obtained precipitate was filtered. In case precipitate was not formed, the mixture was extracted with ethyl acetate (3 5 ml), organic phase dried with MgSO4 and the solvent evaporated. The product was obtained after recrystallization with ethanol or purification with column chromatography (n-hexane/EtOAc).
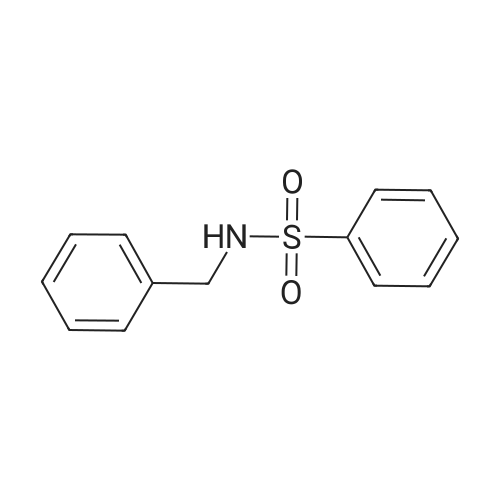
N-benzyl-N-(perfluoropyridin-4-yl)benzenesulfonamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
70%
With potassium carbonate In acetonitrile for 12h; Reflux;
4.1. Reaction of sulfonamides with perhalopyridines; general procedure
General procedure: A mixture of sulfonamide 3 or sulfamide 7 (1 mmol) and perhalopyridine 4 (1 mmol) in the presence of K2CO3 (1 mmol) in CH3CN (5 mL) was stirred at reflux for 12 h. The reaction mixture was poured on 10 mL water and extracted with chloroform (2 × 30 mL), then organic phase dried over MgSO4 and solvent evaporated. The pure product was obtained after washing with ethanol.
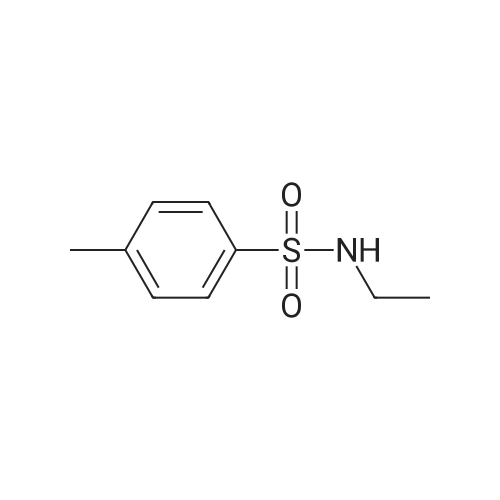
N-ethyl-4-methyl-N-(perfluoropyridin-4-yl)benzenesulfonamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
85%
With potassium carbonate In acetonitrile for 12h; Reflux;
4.1. Reaction of sulfonamides with perhalopyridines; general procedure
General procedure: A mixture of sulfonamide 3 or sulfamide 7 (1 mmol) and perhalopyridine 4 (1 mmol) in the presence of K2CO3 (1 mmol) in CH3CN (5 mL) was stirred at reflux for 12 h. The reaction mixture was poured on 10 mL water and extracted with chloroform (2 × 30 mL), then organic phase dried over MgSO4 and solvent evaporated. The pure product was obtained after washing with ethanol.
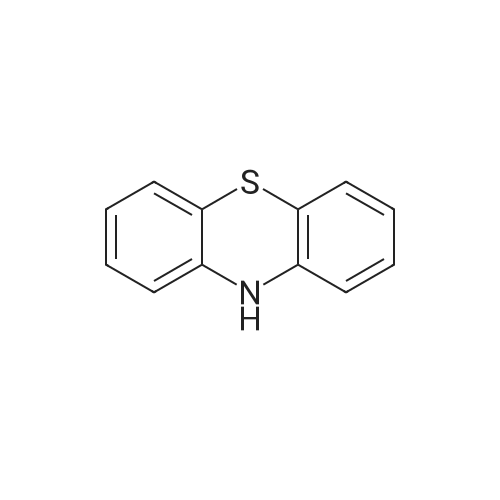
10-(perfluoropyridin-4-yl)-10H-phenothiazine[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
92%
Stage #1: 10H-phenothiazine With potassium phosphate for 1h; Sealed tube;
Stage #2: Pentafluoropyridine In acetonitrile at 60℃; for 24h; Inert atmosphere;
3.2.1. General Procedure A for the Reaction of Phenothiazines with Polyfluoroarenes
General procedure: Phenothiazine derivatives (1.0 mmol) and base (4.0 mmol, 4.0 eq) were placed in ascrew-capped test tube and dried under vacuum for 1 h. After backfilling with N2, solvent(10 mL) and polyfluoroarenes (2.1 mmol, 2.1 eq) were added in this order. The reactionmixture was stirred at 60 C for 24 h. The reaction was quenched with water (50 mL),and the mixture was transferred to a separatory funnel containing diethyl ether (50 mL).The organic layer was separated, and the aqueous layer was extracted with diethyl ether(2 20 mL). The combined organic fractions were washed with brine (50 mL), dried overNa2SO4, and all volatiles were removed under vacuum. The residue was purified by flashcolumn chromatography (SiO2) to yield the corresponding 10-phenylphenothiazine (PTH)derivatives.
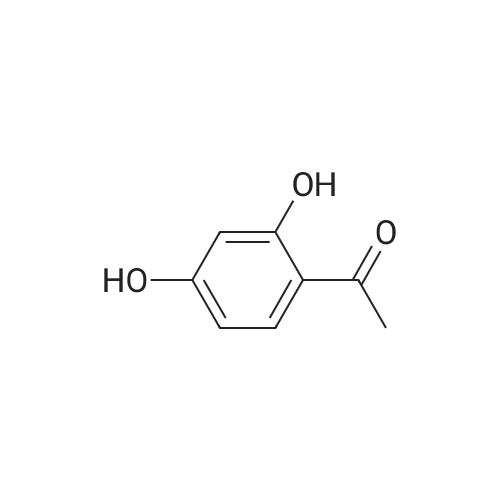
1-(2,4-bis((perfluoropyridin-4-yl)oxy)phenyl)ethanone[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
74%
With potassium carbonate In acetonitrile at 25℃; for 12h;
1-(2,4-bis((Perfluoropyridin-4-yl)oxy)phenyl)ethanone (2)
To a stirred suspension of 1-(2,4-dihydroxyphenyl)ethanone (10, 1 eq., 1 mmol) and anhydrous K2CO3 (2.5 eq., 2.5 mmol)in ACN (5 mL), pentafluoropyridine (2.5 eq., 2.5 mmol) was added. The reaction mixture was stirred at 25 C for 12 h, quenched with HCl (1 M, 10 mL), and the aqueous phase wasextracted with EtOAc (3 5 mL). The combined organic phases were washed with brine(5 mL), dried over Na2SO4, and evaporated under reduced pressure. The crude residuewas purified by silica gel column chromatography (Pet.Et./DCM 2:1) to afford the titlecompound (74% yield) as a white solid. 1H NMR (300 MHz, CDCl3) d 7.95 (d, J = 8.7 Hz,1H, C6-H), 6.89 (dd, J = 8.7, 2.3 Hz, 1H, C5-H), 6.77 (d, J = 2.3 Hz, 1H, C3-H), 2.64 (s, 3H,CH3). 19F NMR (282 MHz, CDCl3) d 85.04-90.34 (m), 150.28-158.26 (m). ESI-MS,m/z [M+H]+ 451.






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science






 HazMat Fee +
HazMat Fee +

 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping